





























IV. Sự thoái hóa
GAGs được thoái hóa trong các lysosomes, lysosomes là nơi chứa các enzyme thủy phân mà hoạt động nhất ở pH khoảng 5. Vì thế, các enzymes này được gọi là các acid hydrolases. Sự tối ưu ở pH thấp là một cơ chế bảo vệ mà giúp ngăn cản enzymes không phá hủy tế bào khi chúng thoát ra vào bên trong bào tương, nơi mà có pH trung tính. Thời gian bán hủy của GAGs thay đổi từ vài phút đến vài tháng và tùy theo loại GAG và vị trí của nó trong cơ thể.
A. GAGs và sự thực bào
Bởi vì GAGs là các hợp chất ngoại bào và trên bề mặt tế bào nên chúng phải được bao lấy bởi sự ấn lõm của màng tế bào (sự thực bào [phagocytosis]), hình thành nên một túi mà ở bên trong đó là các GAGs sẽ bị thoái hóa. Túi này sau đó sẽ hợp với một lysosome, hình thành nên một túi tiêu hóa đơn mà trong đó GAGs được thoái hóa một cách hiệu quả.
B. Sự thoái hóa của lysosome
Sự thoái hóa lysosome của GAGs cần một lượng lớn các acid hydrolases cho sự tiêu hóa hoàn toàn. Đầu tiên, các chuỗi polysaccharide được phân tách bởi các endoglycosidases, tạo thành các oligosaccharides. Sự thoái hóa nhiều hơn nữa của các oligosaccharides xảy ra một cách tuần tự từ đầu không khử của mỗi chuỗi, nhóm cuối cùng (sulfate hoặc đường) được thêm trong suốt quá trình tổng hợp trở thành nhóm đầu tiên bị loại bỏ, bởi hoạt động của các sulfatases hay exoglycosidases. Các ví dụ của một số trong số các enzymes này và các liên kết mà chúng thủy phân được thể hiện trong Hình 12. (Chú ý rằng endo- và exoglycosidases cũng liên quan đến sự thoái hóa của lysosome đối với glycoproteins và glycolipids. Các sự thiếu hụt trong các enzymes này gây ra sự tích tụ của các carbohydrates được thoái hóa một phần, gây ra tổn thương mô).
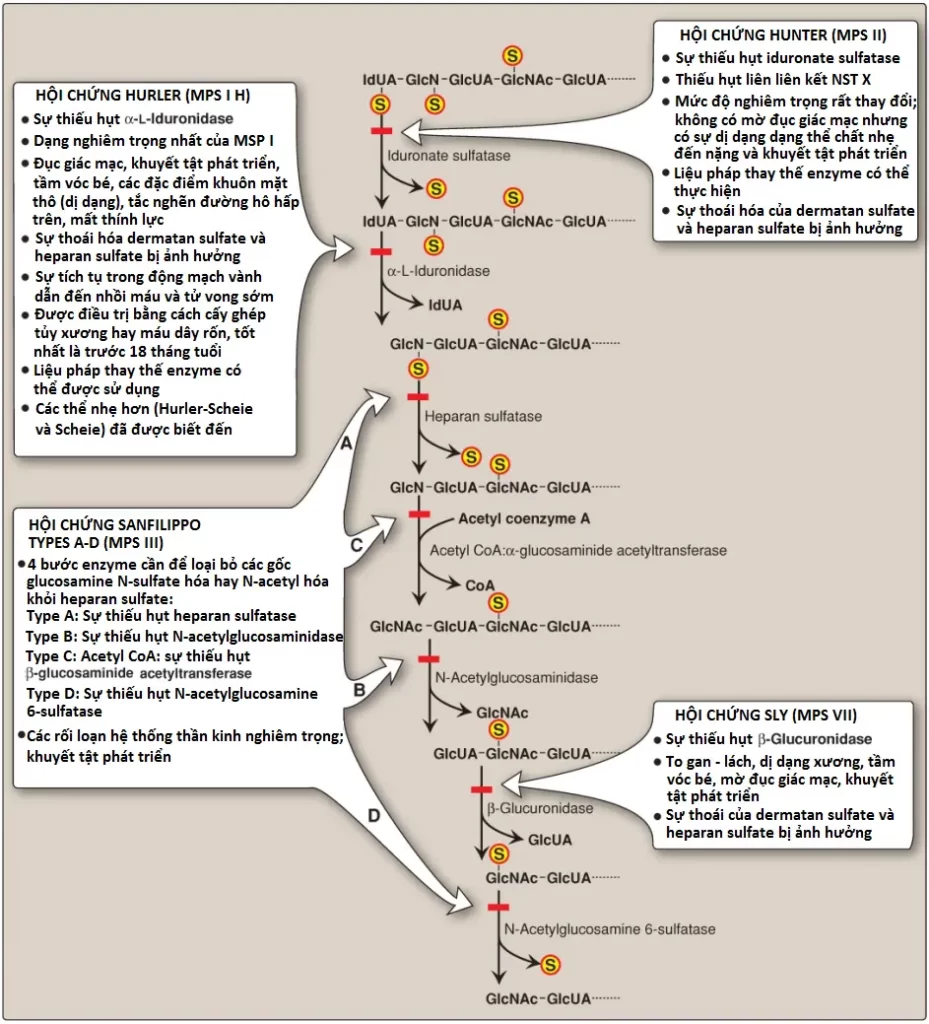
Nhiều sự thiếu hụt sulfatase (bệnh Austin) là một bệnh tích trữ của lysosome hiếm gặp mà trong đó tất cả các sulfatases thì không có chức năng bởi một khiếm khuyết trong sự hình thành của formylglycine, một dẫn xuất amino acid cần thiết tại vị trí hoạt động để hoạt động của enzyme có thể xảy ra.
V. Mucopolysaccharidoses
Mucopolysaccharidoses là các bệnh di truyền (xuất hiện với tỷ lệ xấp xỉ 1:25,000 đứa trẻ sinh ra sống sót) được gây ra bởi một sự thiếu hụt của bất kỳ một trong số các hydrolases của lysosome nào mà bình thường liên quan đến sự thoái hóa của heparan sulfate, dermatan sulfate và/hoặc keratin sulfate (tóm tắt trong Hình 12). Chúng là các rối loạn tiến triển được đặc trưng bởi sự tích tụ trong lysosome của các GAGs trong nhiều mô khác nhau, gây ra nhiều triệu chứng, như biến dạng xương và dịch ngoại bào, và khuyết tật trí tuệ. Tất cả đều là các rối loạn di truyền lặn trên NST thường trừ hội chứng Hunter, là có di truyền liên kết với NST X.
Các trẻ đồng hợp tử đối với bất kỳ một trong số các bệnh này thì sẽ hoàn toàn bình thường lúc sinh ra và sau đó tình trạng dần dần trở nên xấu đi. Trong các tình trạng thiếu hụt nghiêm trọng thì tử vong sẽ xảy ra ở thời thơ ấu. Hiện tại thì không có phương pháp cứu chữa chúng. Sự thoái hóa GAGs của lysosome không hoàn toàn sẽ gây ra sự xuất hiện của các oligosaccharides trong nước tiểu. Các mảnh oligosaccharides này có thể được sử dụng để chẩn đoán mucopolysaccharidosis đặc hiệu bằng cách xác định cấu trúc xuất hiện trên đầu không khử của oligosaccharide, bởi vì gốc đó sẽ đại diện cho cơ chất của enzyme bị thiếu hụt. Chẩn đoán được xác nhận bằng cách đo mức hydrolases lysosome của tế bào bệnh nhân. Cấy ghép tủy xương và cấy ghép máu dây rốn, trong đó các đại thực bào được cấy sẽ sản xuất ra các enzymes mà giúp thoái hóa GAGs, được sử dụng để điều trị các hội chứng Hurler và Hunter với mức độ thành công giới hạn. Liệu pháp thay thế enzyme thì phù hợp với cả hai hội chứng nhưng không ngăn ngừa được tổn thương thần kinh.
VI. Tổng quan về glycoproteins
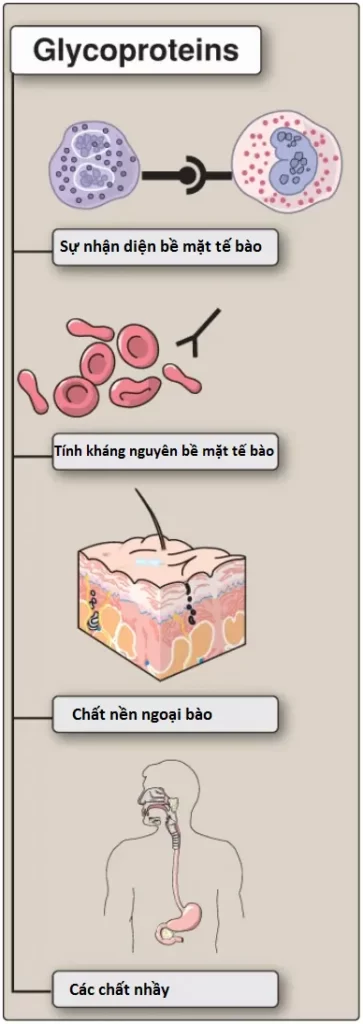
Các glycoproteins là các proteins mà các oligosaccharides (glycans) sẽ được liên kết cộng hóa trị với chúng; sự glycosyl hóa (glycosylation) là sự chỉnh sửa phổ biến nhất sau dịch mã của các proteins. (Chú ý: Sự bổ sung không có xúc tác enzyme của carbohydrate vào các proteins được gọi là sự đường hóa [glycation]). Các glycoproteins chứa các lượng carbohydrate rất thay đổi nhưng thường thì ít hơn nhiều so với của các proteoglycans. Ví dụ, glycoprotein immunoglobulin G (IgG) chứa dưới 4% khối lượng là carbohydrate, ngược lại, proteoglycan aggrecan chứa trên 80%. Trong các glycoproteins, glycan thì tương đối ngắn, thường là 2 đến 10 gốc đường, thường là phân nhánh thay vì là dạng thẳng; và có thể là có hoặc không tích điện âm. Các glycoproteins liên kết màng tham gia vào nhiều hiện tượng của tế bào, bao gồm sự nhận diện bề mặt tế bào của các tế bào khác, hormones và viruses, tính kháng nguyên của bề mặt tế bào (như các kháng nguyên nhóm máu) và khi là các thành phần của dịch ngoại bào và của các chất nhầy của đường tiêu hóa và đường niệu – dục thì chúng đóng vai trò như là các chất bôi trơn sinh học có tác dụng bảo vệ. Ngoài ra, hầu hết tất cả các proteins hình cầu xuất hiện trong huyết tương người là các glycoproteins, mặc dù albumins là một ngoại lệ. Hình 13 tổng hợp một số chức năng của glycoproteins.
VII. Cấu trúc oligosaccharide
Các thành phần oligosaccharide (glycan) của glycoproteins nhìn chung là các heteropolymers phân nhánh bao gồm chủ yếu là D-hexoses, với sự bổ sung trong một số trường hợp là neuraminic acid (một nonose) và L-fucose, một 6-deoxyhexose.
A. Liên kết carbohydrate-protein
Glycan có thể kết nối với protein qua một N- hoặc O-glycoside. Trong trường hợp trước, chuỗi đường được nối với nhóm amide của một chuỗi bên asparagine và trong trường hợp sau, là với nhóm hydroxyl của một chuỗi bên serine và threonine. Trong trường hợp của collagen, có một liên kết O-glycoside giữa galactose hay glucose và nhóm hydroxyl của hydroxylysine.
B. N- và O- oligosaccharides
Một glycoprotein có thể chứa chỉ một loại liên kết glycoside (liên kết N hoặc O) hoặc có thể có cả hai loại liên kết bên trong cùng một phân tử.
1. Liên kết O: Các glycans liên kết O có thể có một hoặc nhiều hơn các loại đường khác nhau được sắp xếp theo kiểu thẳng hoặc phân nhánh. Nhiều trong số đó thì được tìm thấy trong các glycoproteins ngoại bào hay là các thành phần glycoprotein màng. Ví dụ, các oligosaccharides liên kết O trên bề mặt của các tế bào hồng cầu giúp cung cấp các sự xác định nhóm máu ABO. Nếu như đường tận cùng trên glycan là GalNAc, nhóm máu là A. Nếu như nó là galactose, nhóm máu là B. Nếu như không có cả GalNAc hay galactose thì nhóm máu là O.
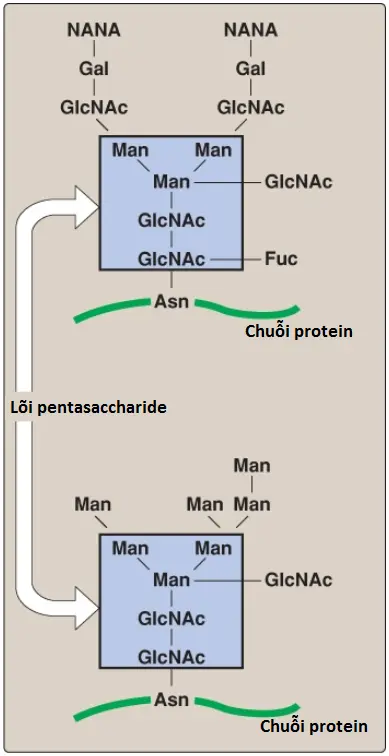
2. Liên kết N: Các glycans liên kết N rơi vào hai loại lớn: các oligosaccharides phức tạp và các oligosaccharides nhiều mannose. Cả hai đều có cùng lõi pentasaccharide được thể hiện trong Hình 14, nhưng các oligosaccharides phức tạp chứa một nhóm phong phú các đường bổ sung, ví dụ, GlcNAc, GalNAc, L-fucose và NANA, ngược lại, các oligosaccharides nhiều mannose chứa chủ yếu mannose.
VII. Sự tổng hợp glycoproteins
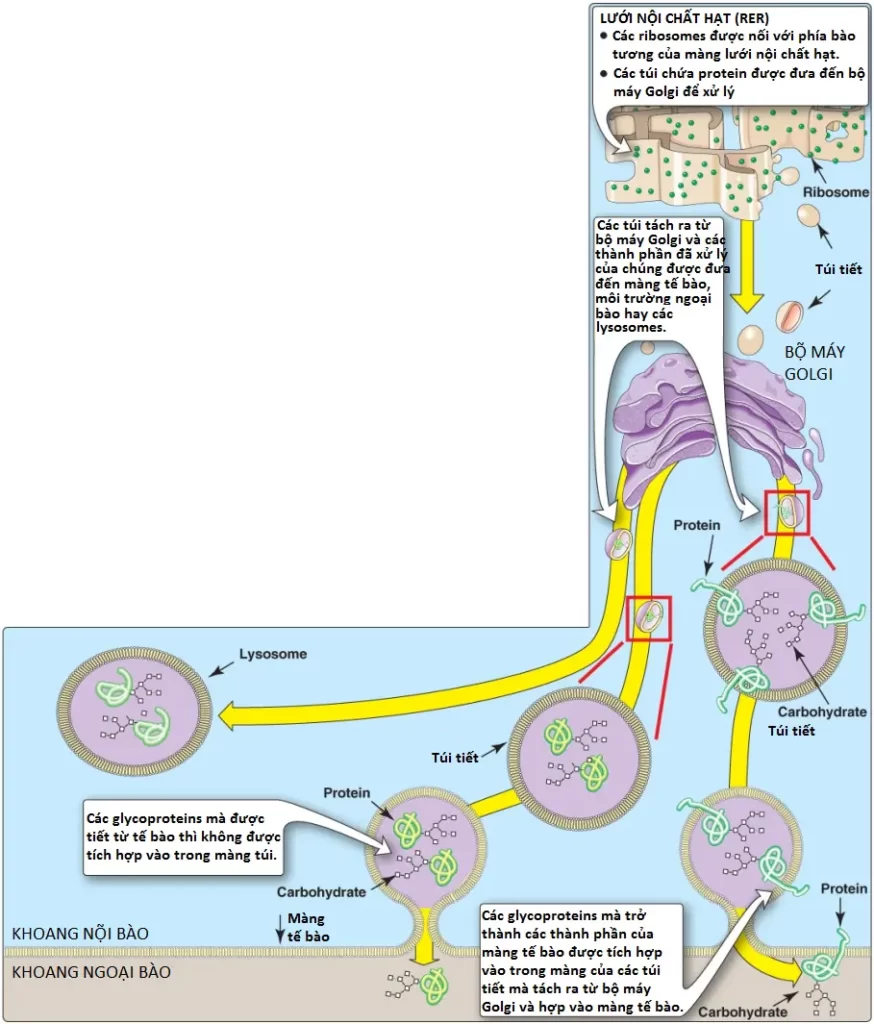
Các protein mà có số phận thực hiện chức năng trong bào tương thì được tổng hợp trên các ribosomes bào tương tự do. Tuy nhiên, các proteins, bao gồm các glycoproteins mà có số phận ở các màng tế bào, lysosomes hoặc để bài tiết ra ngoài tế bào thì sẽ được tổng hợp trên các ribosomes mà bám vào lưới nội chất. Các proteins này chứa các trình tự tín hiệu đặc hiệu mà đóng vai trò như là các địa chỉ phân tử, điều hướng các proteins đến các đích đến thích hợp của nó. Một trình tự kỵ nước đầu tận N ban đầu sẽ hướng các protein này đến lưới nội chất, cho phép chuỗi polypeptide đang phát triển được phóng xuất vào trong lòng lưới. Các proteins sau đó được vận chuyển qua các túi tiết đến bộ máy Golgi, nơi đóng vai trò như là một trung tâm phân loại (Hình 15). Trong bộ máy Golgi, các glycoproteins mà được bài tiết khỏi tế bào hay hướng đến lysosomes thì sẽ được đóng gói vào trong các túi mà sẽ hợp với màng tế bào hay màng lysosome và phóng xuất các thành phần của nó. Những glycoproteins mà có số phận trở thành các thành phần của màng tế bào thì được tích hợp vào trong màng bộ máy Golgi, thành phần mà sẽ tách ra, hình thành nên các túi mà thêm các glycoproteins liên kết màng của nó đến màng tế bào và được định hướng với phần carbohydrate hướng về phía ngoài tế bào (xem Hình 15).
A. Các thành phần carbohydrate
Các tiền thân của các thành phần carbohydrate của các glycoproteins là các đường nucleotides mà bao gồm UDP-glucose, UDP-galactose, UDP-GlcNAc và UDP-GalNAc. Ngoài ra, guanosine diphosphate (GDP)-mannose, GDP-L-fucose (được tổng hợp từ GDP-mannose) và CMP-NANA có thể cho các đường đến chuỗi đang phát triển. Khi NANA có tính acid xuất hiện thì oligosaccharide có một sự tích điện âm ở pH sinh lý. Các oligosaccharides được liên kết một cách cộng hóa trị đến các chuỗi bên của các amino acids đặc hiệu trong protein, nơi mà cấu trúc ba chiều của protein xác định việc liệu có hay không một amino acid đặc hiệu được glycosyl hóa.
B. Sự tổng hợp glycoprotein liên kết O
Sự tổng hợp các glycoproteins liên kết O thì rất giống với sự tổng hợp của các GAGs. Đầu tiên, protein mà các đường liên kết vào thì được tổng hợp trên lưới nội chất hạt (RER) và phóng xuất vào trong lòng lưới. Sự glycosyl hóa bắt đầu với sự chuyển GalNAc (từ UDP-GalNAc) đến nhóm hydroxyl của một gốc serine hay threonine đặc hiệu. Các glycosyltransferases chịu trách nhiệm cho sự tổng hợp theo từng bước (từ các đường riêng rẽ) của các oligosaccharides thì được liên kết với màng của bộ máy Golgi. Chúng hoạt động theo một thứ tự chuyên biệt mà không sử dụng một khuôn mẫu như cần đối với DNA, ribonucleic acid (RNA) và sự tổng hợp protein mà thay vào đó bằng cách nhận diện cấu trúc thực sự của oligosaccharide đang phát triển như là một cơ chất thích hợp.
C. Sự tổng hợp glycoprotein liên kết N
Sự tổng hợp glycoprotein liên kết N xảy ra trong lòng lưới nội chất hạt và cần sự tham gia của dạng phosphoryl hóa của dolichol (dolichol pyrophosphate), một lipid của màng lưới nội chất hạt (Hình 16). Sản phẩm ban đầu được xử lý trong lưới nội chất hạt và bộ máy Golgi.
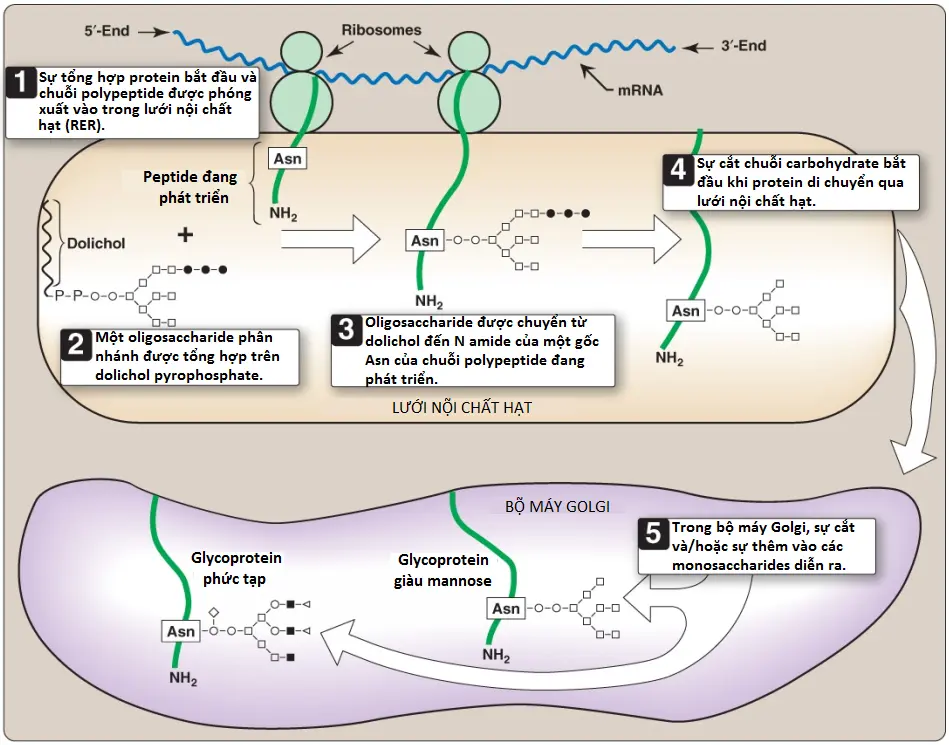
1. Sự tổng hợp oligosaccharide liên kết dolichol: Giống với các glycoproteins liên kết O, protein được tổng hợp trên lưới nội chất hạt và đi vào trong lòng ống lưới. Tuy nhiên, nó không được glycosyl hóa với các đường riêng rẽ. Thay vào đó, một oligosaccharide liên kết lipid được cấu trúc trước tiên. Quá trình này bao gồm dolichol, một lipid màng lưới nội chất hạt được tạo thành từ một trung gian của sự tổng hợp cholesterol nối qua một liên kết pyrophosphate với một oligosaccharide chứa GlcNAc, mannose và glucose. Các đường được thêm một cách tuần tự vào dolichol bởi glycosyltransferases liên kết màng là GlcNAc đầu tiên, theo sau bởi mannose và glucose (xem Hình 16). Toàn bộ oligosaccharide 14-đường sau đó được chuyển từ dolichol đến nitrogen amide của một gốc asparagine trong protein được glycosyl hóa bởi một protein-oligosaccharide transferase xuất hiện trong RER. (Chú ý: Kháng sinh Tunicamycin ức chế sự glycosyl hóa liên kết N).
Các rối loạn di truyền của sự glycosyl hóa (congenital disorders of glycosylation – CDG) là các hội chứng mà được gây ra chủ yếu bởi các khiếm khuyết trong sự glycosyl hóa liên kết N của các proteins, hoặc lắp ráp oligosaccharide (type I) hoặc xử lý oligosaccharide (type II).
2. Quá trình xử lý oligosaccharide liên kết N: Sau khi được thêm vào protein, oligosaccharide liên kết N được xử lý bằng cách loại bỏ các gốc mannosyl và glucosyl đặc hiệu khi glycoprotein di chuyển qua lưới nội chất hạt. Cuối cùng, các chuỗi oligosaccharide được hoàn thành trong bộ máy Golgi bằng cách thêm nhiều loại đường khác nhau (ví dụ, GlcNAc, GalNAc, các mannoses thêm vào và sau đó là fucose hoặc NANA đóng vai trò là các nhóm tận cùng) để sản xuất ra một glycoprotein phức tạp. Ngoài ra, chúng có thể không được xử lý thêm nữa, để lại các chuỗi chứa mannose phân nhánh trong một glycoprotein chứa nhiều mannose (xem Hình 16). Số phận cuối cùng của các glycoproteins liên kết N thì giống với các glycoproteins liên kết O (ví dụ, chúng có thể được giải phóng khỏi tế bào hoặc trở thành một phần của màng tế bào. Ngoài ra, các glycoproteins liên kết N có thể được điều hướng đến các lysosomes).
3. Các enzyme của lysosome: Các glycoproteins liên kết N được xử lý trong bộ máy Golgi có thể được phosphoryl hóa trên carbon thứ 6 của một hoặc nhiều gốc mannosyl. UDP-GlcNAc cung cấp phosphate trong một phản ứng được xúc tác bởi một phosphotransferase. Các thụ cảm thể (receptors), nằm trên màng của bộ máy Golgi, liên kết với các gốc mannose 6-phosphate (M6P) của các proteins này, thành phần mà sau đó được bọc vào trong các túi và được gửi đến các lysosomes (Hình 17).
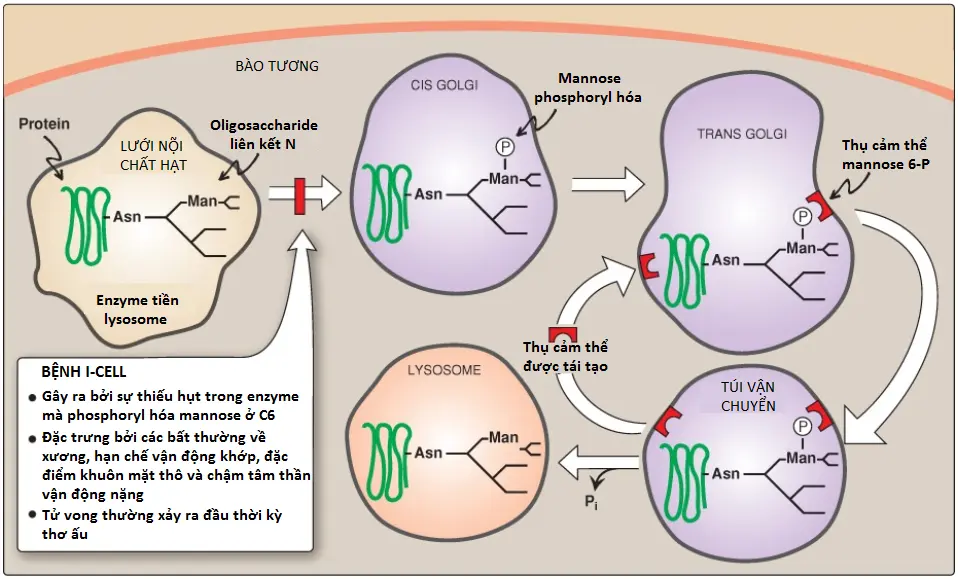
Ứng dụng lâm sàng 2: Bệnh I-Cell
Bệnh I-Cell là một bệnh tích trữ của lysosome hiếm gặp mà được đặt tên theo các thể vùi lớn được quan sát thấy trong các tế bào của các bệnh nhân mắc bệnh. GlcNAc phosphotransferases thì thiếu hụt và mannose 6-phosphate thì không được tạo ra trên các proteins đến lysosomes. Thiếu M6P trên các gốc amino acid làm cho tiền thân các acid hydrolase được đưa đến màng bào tương và được bài tiết ra ngoài thay vì là đi đến lysosomes. Kết quả là các acid hydrolase không có mặt tại lysosomes và các cơ chất đại phân tử của các enzymes tiêu hóa này tích tụ bên trong các lysosomes, tạo ra các thể vùi mà gây ra sự rối loạn. Ngoài ra, nồng độ của cao của các enzyme lysosomes được tìm thấy trong huyết tương và nước tiểu của bệnh nhân cho thấy rằng quá trình hướng mục tiêu đến các lysosomes bị khiếm khuyết.
Bệnh I-Cell được đặc trưng bởi các bất thường xương, vận động khớp hạn chế, các đặc điểm khuôn mặt thô (dị dạng) và chậm tâm thần vận động (psychomotor impairment [retardation]) nghiêm trọng. Bởi vì bệnh I-Cell có các đặc điểm chung với các mucopolysaccharidoses và sphingolipidoses nên nó được gọi là mucolipidosis (ML II). Hiện tại, không có cách cứu chữa và tử vong do các biến chứng tim – phổi thường xảy ra ở đầu thời thơ ấu. Bệnh đa loạn dưỡng giả Hurler (pseudo-Hurler polydystrophy) (ML III) là một dạng mucolipidosis ít nghiêm trọng hơn của bệnh I-Cell, trong đó, phosphotransferase duy trì một số hoạt tính enzyme còn lại và nó tương tự với một dạng nhẹ của hội chứng Hurler về mặt triệu chứng.
IX. Sự thoái hóa glycoprotein của lysosome
Sự thoái hóa của các glycoproteins thì tương tự với của GAGs. Mỗi acid hydrolases của lysosome thì nhìn chung là đặc hiệu cho sự loại bỏ một thành phần của glycoprotein. Chúng chủ yếu là các exoenzymes mà giúp loại bỏ các nhóm tương ứng theo thứ tự ngược lại với sự tích hợp ban đầu (last on, first off [nghĩa là tích hợp vào sau thì sẽ tách ra trước]). Nếu như bất cứ enzyme thoái hóa nào bị thiếu đi thì sự thoái hóa bởi các exoenzymes khác không thể tiếp tục.
Một nhóm các bệnh di truyền lặn trên NST thường rất hiếm được gọi là các bệnh tích trữ glycoprotein (oligosaccharidoses), được gây ra bởi một sự thiếu hụt của bất kỳ một trong số các enzymes thoái hóa nào, gây ra sự tích tụ của các cấu trúc bị thoái hóa một phần trong các lysosomes. Ví dụ, α-mannosidosis type 3 là một sự thiếu hụt nặng, tiến triển, gây tử vong của enzyme α-mannosidase. Biểu hiện thì tương tự với hội chứng Hurler nhưng sự suy giảm miễn dịch cũng được quan sát thấy. Các mảnh oligosaccharide giàu mannose xuất hiện trong nước tiểu. Chẩn đoán bởi xét nghiệm hoạt động enzyme.
X. Tóm tắt bài viết
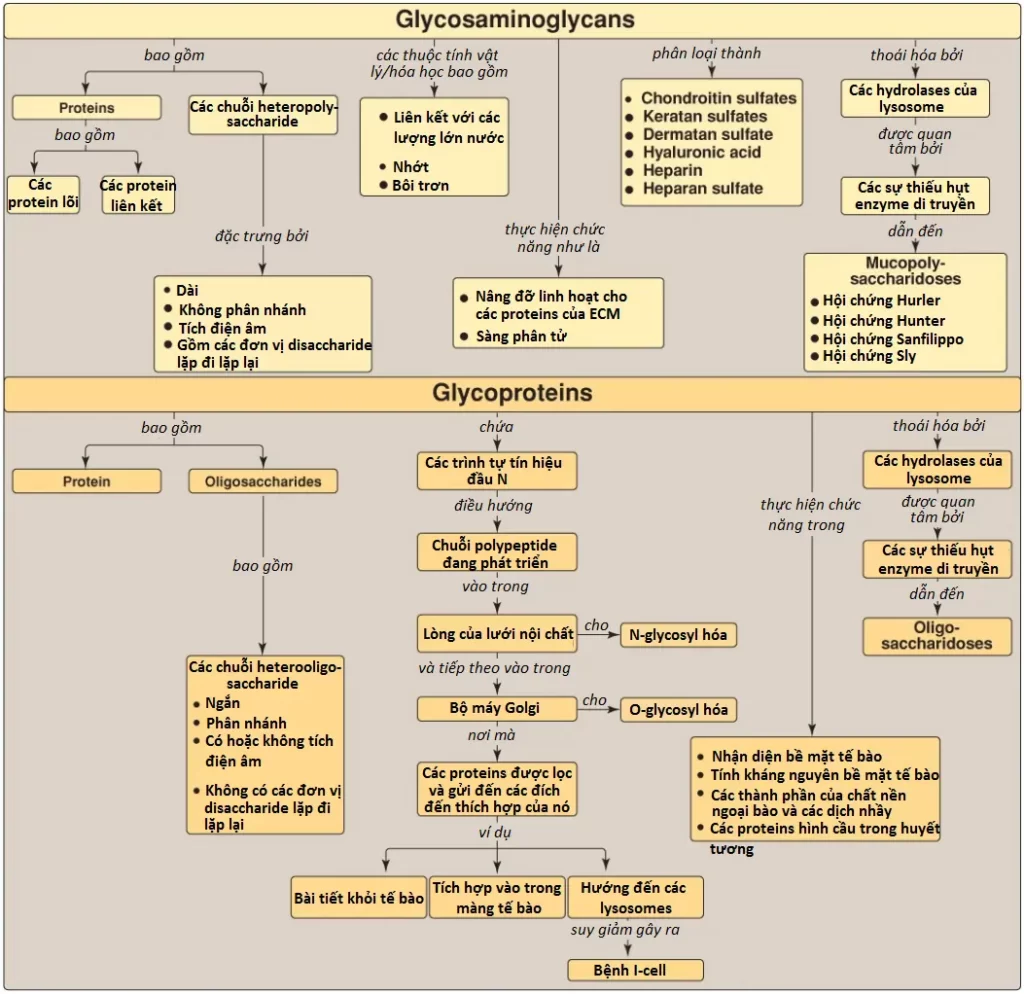
- GAGs được tổng hợp trong bộ máy Golgi dưới dạng các chuỗi heteropolysaccharide dài, tích điện âm, không phân nhánh nhìn chung là bao gồm các đơn vị disaccharide (đường acid-đường amino)n.
- Đường amino thì là D-glucosamine hoặc D-galactosamine và đường acid là D-glucuronic acid hoặc epimer C-5 của nó là L-iduronic acid.
- GAGs liên kết với nước, bằng cách đó, tạo thành chất nền dạng gel mà hình thành nên phần cơ bản của chất nền cơ thể và các thuộc tính bôi trơn của các sự bài tiết nhầy.
- Có 6 loại GAGs chính: chondroitin 4- và 6-sulfates, keratan sulfate, dermatan sulfate, heparin, heparan sulfate và hyaluronic acid.
- Tất cả các GAGs, trừ hyaluronic acid là được tìm thấy có liên kết cộng hóa trị với một protein lõi, hình thành nên các monomers proteoglycan. Nhiều monomers proteoglycan liên kết với một phân tử hyaluronic acid để hình thành nên các tập hợp proteoglycan.
- Các proteoglycans hoàn thành được tiết vào trong chất nền ngoại bào hoặc vẫn liên kết với mặt ngoài của các tế bào.
- GAGs bị thoái hóa bởi các acid hydrolases của lysosome. Một sự thiếu hụt trong bất kỳ loại hydrolases nào sẽ gây ra một tình trạng bệnh được gọi là mucopolysaccharidosis, trong đó GAGs tích tụ trong các mô, gây ra các triệu chứng như các biến dạng xương và chất nền ngoại bào và khuyết tật trí tuệ. Các ví dụ cho các bệnh di truyền này bao gồm các hội chứng Hunter (liên kết NST X) và Hurler.
- Các glycoproteins được tổng hợp trong lưới nội chất hạt và bộ máy Golgi và là các proteins mà liên kết một cách cộng hóa trị với các oligosaccharides (glycans).
- Các glycoproteins liên kết màng tham gia vào sự nhận diện bề mặt tế bào, tính kháng nguyên của bề mặt tế bào và khi là các thành phần của chất nền ngoại bào và của các chất nhầy của đường tiêu hóa và đường niệu dục thì chúng đóng vai trò như là các chất bôi trơn sinh học. Hầu hết tất cả các proteins hình cầu xuất hiện trong huyết tương người là các glycoproteins.
- Các tiền thân của các thành phần carbohydrate của glycoproteins là các đường nucleotide. Các glycoproteins liên kết O được sản xuất trong bộ máy Golgi bởi sự chuyển lần lượt của các đường từ các chất mang nucleotide của chúng đến nhóm hydroxyl của một gốc Ser (serine) hay Thr (threonine) trong protein. Các glycoproteins liên kết N được tạo ra bằng cách chuyển một oligosaccharide hình thành từ trước từ chất mang lipid của màng nội chất hạt là dolichol pyrophosphate, đến nitrogen amide của một gốc Asn (asparagine) trong protein. Chúng chứa các lượng mannose khác nhau.
- Một sự thiếu hụt trong N-acetylglucosamine phosphotransferase mà phosphoryl hóa các gốc mannose ở carbon thứ 6 trong các enzymes glycoprotein liên kết N đến lysosomes gây ra bệnh I-cell.
- Các glycoproteins bình thường được thoái hóa trong các lysosomes bởi các acid hydrolases. Một sự thiếu hụt của bất kỳ một trong số các enzyme này sẽ gây ra một bệnh tích trữ glycoprotein của lysosome, tạo ra sự tích tụ của các glycoproteins được thoái hóa một phần trong lysosome và gây ra nhiều triệu chứng bao gồm biến dạng xương và khuyết tật trí tuệ.
Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/profile.php?id=61550892771585
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/glycosaminoglycans-proteoglycans-va-glycoproteins-phan-1/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!







{kind=link}