





























IV. Sự phân phối thuốc
E. Thể tích phân bố
Thể tích phân bố biểu kiến, Vd, được định nghĩa là thể tích chất lỏng cần thiết để chứa toàn bộ thuốc trong cơ thể ở cùng nồng độ được đo trong huyết tương. Nó được tính bằng cách chia liều cuối cùng đi vào tuần hoàn hệ thống cho nồng độ huyết tương tại thời điểm không (C0).
Vd = Lượng thuốc trong cơ thể/C0
Mặc dù Vd không có cơ sở sinh lý hoặc vật lý, nhưng có thể hữu ích khi so sánh sự phân bố của thuốc với thể tích của các khoang nước trong cơ thể.
1. Phân bố vào các khoang nước trong cơ thể
Khi một loại thuốc đi vào cơ thể, nó có khả năng phân bố vào bất kỳ một trong ba khoang nước riêng biệt về mặt chức năng của cơ thể hoặc bị cô lập tại một vị trí tế bào.
a. Khoang huyết tương
Nếu một loại thuốc có trọng lượng phân tử cao hoặc liên kết nhiều với protein, thì nó quá lớn để đi qua các khe nối của mao mạch và do đó, bị giữ lại hiệu quả trong khoang huyết tương (mạch máu). Do đó, nó có Vd thấp, xấp xỉ thể tích huyết tương hay khoảng 4 L ở một cá nhân nặng 70 kg. Heparin (xem loạt bài viết sau) cho thấy loại phân bố này.
b. Dịch ngoại bào
Nếu một loại thuốc có trọng lượng phân tử thấp nhưng ưa nước, nó có thể đi qua các khe nối nội mô của mao mạch vào dịch kẽ. Tuy nhiên, các loại thuốc ưa nước không thể di chuyển qua màng lipid của tế bào để vào dịch nội bào. Do đó, các loại thuốc này phân phối vào một thể tích là tổng thể tích huyết tương và dịch kẽ, mà cùng nhau tạo thành dịch ngoại bào (khoảng 20% trọng lượng cơ thể hay 14 L ở một cá nhân nặng 70 kg). Kháng sinh aminoglycoside (xem loạt bài viết sau) cho thấy loại phân phối này.
c. Tổng lượng nước trong cơ thể
Nếu một loại thuốc có trọng lượng phân tử thấp và có đủ tính ưa mỡ, nó có thể di chuyển vào mô kẽ qua các khe nối và đi qua màng tế bào vào dịch nội bào. Các loại thuốc này phân phối vào một thể tích khoảng 60% trọng lượng cơ thể hoặc khoảng 42 L ở một cá nhân nặng 70 kg. Ethanol thể hiện Vd biểu kiến này. [Lưu ý: Nhìn chung, Vd lớn hơn biểu thị sự phân bố lớn hơn vào các mô; Vd nhỏ hơn biểu thị sự giới hạn trong huyết tương hoặc dịch ngoại bào.]
2. Xác định Vd
Thực tế là độ thanh thải thuốc thường là một quá trình bậc nhất cho phép tính toán Vd. Bậc nhất có nghĩa là một phần không đổi của thuốc được đào thải trên một đơn vị thời gian. Quá trình này có thể được phân tích dễ dàng nhất bằng cách vẽ biểu đồ logarit của nồng độ thuốc trong huyết tương (Cp) theo thời gian (Hình 1). Nồng độ thuốc trong huyết tương có thể được ngoại suy ngược về thời điểm không (thời điểm tiêm tĩnh mạch nhanh) trên trục Y để xác định C0, đây là nồng độ thuốc sẽ đạt được nếu giai đoạn phân bố xảy ra ngay lập tức. Điều này cho phép tính toán Vd như sau:
Vd = Liều lượng/C0
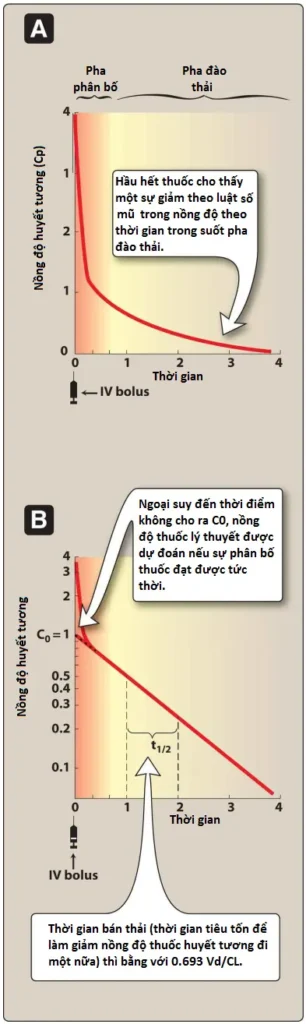
Ví dụ, nếu tiêm 10 mg thuốc vào bệnh nhân và nồng độ trong huyết tương được ngoại suy ngược về thời điểm không và C0 = 1 mg/L (từ đồ thị trong Hình 1B), thì Vd = 10 mg/1 mg/L = 10 L.
3. Ảnh hưởng của Vd đến thời gian bán thải của thuốc
Vd có ảnh hưởng quan trọng đến thời gian bán thải của thuốc, vì quá trình đào thải thuốc phụ thuộc vào lượng thuốc được đưa đến gan hoặc thận (hoặc các cơ quan khác nơi diễn ra quá trình chuyển hóa) trên một đơn vị thời gian. Quá trình đưa thuốc đến các cơ quan đào thải không chỉ phụ thuộc vào lưu lượng máu mà còn phụ thuộc vào tỷ lệ thuốc trong huyết tương. Nếu thuốc có Vd lớn, phần lớn thuốc sẽ ở trong không gian ngoại bào và không có sẵn cho các cơ quan bài tiết. Do đó, bất kỳ yếu tố nào làm tăng Vd đều có thể làm tăng thời gian bán thải và kéo dài thời gian tác dụng của thuốc. [Lưu ý: Vd đặc biệt lớn cho thấy thuốc bị cô lập đáng kể trong một số mô hoặc khoang.]
V. Độ thanh thải thuốc thông qua quá trình chuyển hóa
Khi thuốc đi vào cơ thể, quá trình đào thải bắt đầu. Ba con đường đào thải chính là chuyển hóa ở gan, đào thải qua mật và bài tiết qua nước tiểu. [Lưu ý: Đào thải là quá trình loại bỏ thuốc không thể đảo ngược khỏi cơ thể. Quá trình này bao gồm chuyển hóa sinh học (chuyển hóa thuốc) và bài tiết. Bài tiết là quá trình loại bỏ thuốc còn nguyên vẹn khỏi cơ thể.] Cùng nhau, các quá trình đào thải này làm giảm nồng độ thuốc trong huyết tương theo luật số mũ. Nghĩa là, một phần không đổi của thuốc được đào thải trong một đơn vị thời gian nhất định (Hình 1A). Quá trình chuyển hóa tạo ra các sản phẩm có độ phân cực tăng lên, cho phép thuốc được đào thải. Độ thanh thải (clearance – CL) ước tính thể tích máu mà thuốc được đào thải trên một đơn vị thời gian. CL toàn phần là ước tính tổng hợp phản ánh tất cả các cơ chế đào thải thuốc và được tính toán như sau:
CL = 0.693 x Vd / t1/2
trong đó t1/2 là thời gian bán thải, Vd là thể tích phân bố biểu kiến và 0.693 là hằng số logarit tự nhiên. Thời gian bán hủy của thuốc thường được dùng làm thước đo CL của thuốc vì đối với nhiều loại thuốc, Vd là một hằng số.
A. Động học chuyển hóa
1. Động học bậc nhất
Quá trình chuyển hóa thuốc được xúc tác bởi enzymes và hầu hết các phản ứng đều tuân theo động học Michaelis-Menten, trong đó Km là hằng số Michaelis (nồng độ cơ chất ở nửa vận tốc cực đại).
v = Tốc độ chuyển hóa thuốc = Vmax [C]/Km + [C]
Trong hầu hết các trường hợp lâm sàng, nồng độ thuốc, [C], nhỏ hơn nhiều so với hằng số Michaelis, Km, và phương trình Michaelis-Menten được rút gọn thành
v = Tốc độ chuyển hóa thuốc = Vmax [C]/Km
Nghĩa là, tốc độ chuyển hóa và đào thải thuốc tỷ lệ thuận với nồng độ thuốc tự do và động học bậc nhất được quan sát thấy (Hình 2). Điều này có nghĩa là một phần thuốc không đổi được chuyển hóa trên một đơn vị thời gian (tức là với mỗi chu kỳ bán hủy, nồng độ giảm 50%). Động học bậc nhất cũng được gọi là động học tuyến tính.
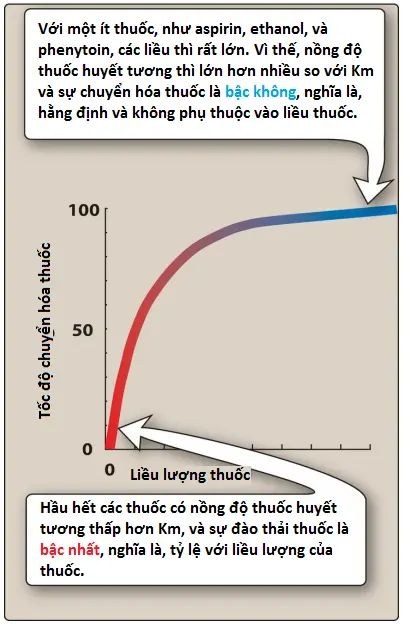
2. Động học bậc không
Với một số ít thuốc, chẳng hạn như aspirin, ethanol và phenytoin, liều lượng rất lớn. Do đó, [C] lớn hơn nhiều so với Km, và phương trình vận tốc trở thành
v = Tốc độ chuyển hóa thuốc = Vmax [C]/[C] = Vmax
Enzyme được bão hòa bởi nồng độ thuốc tự do cao và tốc độ chuyển hóa vẫn không đổi theo thời gian. Đây được gọi là động học bậc không (còn gọi là động học phi tuyến tính). Một lượng thuốc không đổi được chuyển hóa trên một đơn vị thời gian. Tốc độ đào thải là không đổi và không phụ thuộc vào nồng độ thuốc.
B. Phản ứng chuyển hóa thuốc
Thận không thể bài tiết hiệu quả các thuốc thân dầu dễ dàng đi qua màng tế bào và được tái hấp thu ở các ống lượn xa. Do đó, các tác nhân hòa tan trong lipid trước tiên được chuyển hóa thành các chất phân cực hơn (ưa nước) ở gan thông qua hai tập hợp phản ứng chung, được gọi là pha I và pha II (Hình 3).
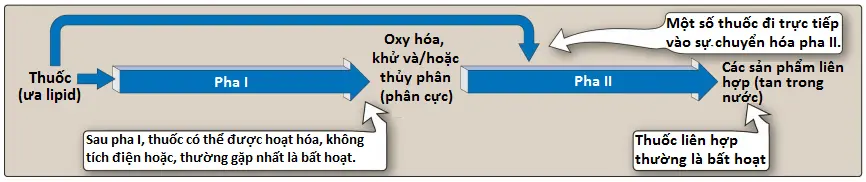
1. Pha I
Phản ứng pha I chuyển đổi các thuốc thân dầu thành các phân tử phân cực hơn bằng cách đưa vào hoặc làm lộ một nhóm chức phân cực, chẳng hạn như –OH hoặc –NH2. Phản ứng pha I thường liên quan đến quá trình khử, oxy hóa hoặc thủy phân. Chuyển hóa pha I có thể tăng, giảm hoặc không ảnh hưởng đến hoạt động dược lý.
a. Phản ứng pha I sử dụng hệ thống P450
Các phản ứng pha I thường xuyên nhất liên quan đến chuyển hóa thuốc được xúc tác bởi hệ thống cytochrome P450 (CYP). Hệ thống P450 rất quan trọng đối với quá trình chuyển hóa nhiều hợp chất nội sinh (như steroid, lipid) và đối với quá trình chuyển hóa sinh học các chất ngoại sinh (thuốc, chất gây ung thư và chất gây ô nhiễm môi trường). CYP là một siêu họ của các isozymes chứa heme nằm trong hầu hết các tế bào, nhưng chủ yếu ở gan và đường tiêu hóa.
[1] Danh pháp
Tên họ được chỉ định bằng số Ả Rập theo sau CYP và chữ cái viết hoa chỉ định phân họ, ví dụ, CYP3A (Hình 4). Một số thứ hai chỉ isozyme cụ thể, như trong CYP3A4.
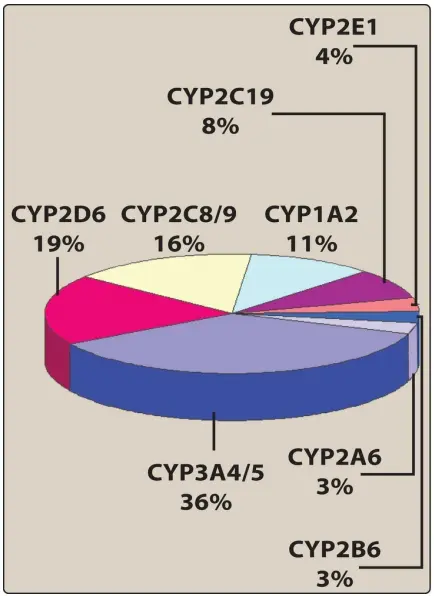
[2] Tính đặc hiệu
Vì có nhiều gene khác nhau mã hóa nhiều loại enzymes, nên có nhiều isoforms P450 khác nhau. Các enzyme này có khả năng biến đổi một số lượng lớn các cơ chất có cấu trúc đa dạng. Ngoài ra, một loại thuốc riêng lẻ có thể là cơ chất cho nhiều hơn một isozyme. Bốn isozyme (CYP3A4/5, CYP2D6, CYP2C8/9 và CYP1A2) chịu trách nhiệm cho phần lớn các phản ứng do P450 xúc tác (Hình 4). Một lượng đáng kể CYP3A4 được tìm thấy trong niêm mạc ruột, đóng góp vào quá trình chuyển hóa lần đầu của các loại thuốc như chlorpromazine và clonazepam.
[3] Biến đổi di truyền
Các enzyme P450 biểu hiện sự biến đổi di truyền đáng kể giữa các cá nhân và nhóm chủng tộc. Sự thay đổi trong hoạt động của P450 có thể làm thay đổi hiệu quả của thuốc và nguy cơ xảy ra các tác dụng phụ. Đặc biệt, CYP2D6 biểu hiện đa hình di truyền. Các đột biến CYP2D6 dẫn đến khả năng chuyển hóa các cơ chất rất thấp. Ví dụ, một số cá nhân không nhận được lợi ích từ thuốc giảm đau opioid codeine, vì họ thiếu enzyme CYP2D6 hoạt hóa thuốc. Các đa hình tương tự đã được mô tả cho phân họ isozyme CYP2C. Ví dụ, clopidogrel có cảnh báo rằng những bệnh nhân là “người chuyển hóa kém” CYP2C19 sẽ có tác dụng chống kết tập tiểu cầu giảm khi dùng thuốc này và nên cân nhắc dùng thuốc thay thế. Clopidogrel là một tiền chất và hoạt động của CYP2C19 là cần thiết để chuyển đổi nó thành chất chuyển hóa có hoạt tính.
[4] Chất cảm ứng CYP
Các enzymes phụ thuộc CYP450 là một mục tiêu quan trọng đối với các tương tác thuốc dược động học. Một số loại thuốc nhất định (ví dụ, phenobarbital, rifampin và carbamazepine) có khả năng cảm ứng các isozyme CYP. Điều này dẫn đến tăng chuyển hóa sinh học của thuốc và có thể dẫn đến giảm đáng kể nồng độ trong huyết tương của các thuốc được chuyển hóa bởi các isozyme CYP này, thường đồng thời mất tác dụng dược lý. Ví dụ, rifampin, một loại thuốc chống lao (xem Chương 32), làm giảm đáng kể nồng độ chất ức chế protease của virus gây suy giảm miễn dịch ở người (HIV) trong huyết tương, do đó làm giảm khả năng ức chế sự sao chép của HIV. Hình 5 liệt kê một số chất cảm ứng quan trọng hơn đối với các isozyme CYP tiêu biểu.
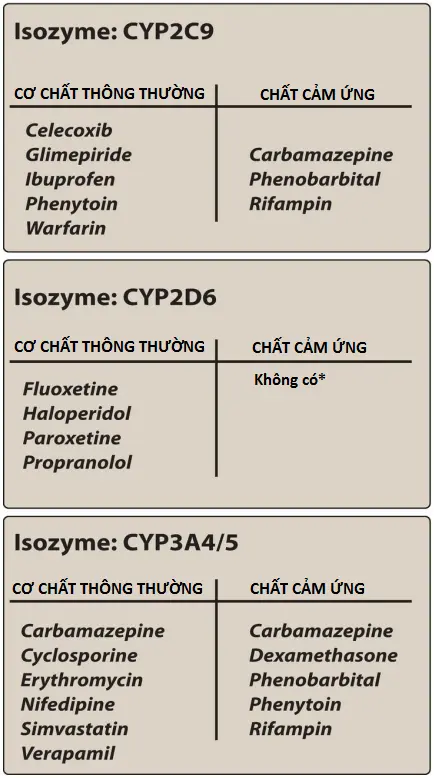
[5] Chất ức chế CYP
Ức chế chuyển hóa thuốc có thể dẫn đến tăng đáng kể nồng độ thuốc trong huyết tương và các tác dụng phụ hoặc độc tính của thuốc. Hình thức ức chế phổ biến nhất là thông qua sự cạnh tranh cho cùng một isozyme. Tuy nhiên, một số loại thuốc có khả năng ức chế các phản ứng mà chúng không phải là cơ chất (ví dụ, ketoconazole), dẫn đến tương tác thuốc. Nhiều loại thuốc ức chế một hoặc nhiều con đường chuyển dạng sinh học phụ thuộc CYP của warfarin. Ví dụ, omeprazole là chất ức chế mạnh ba isozyme CYP tham gia vào quá trình chuyển hóa warfarin. Khi dùng cùng với omeprazole, nồng độ warfarin trong huyết tương tăng lên, dẫn đến tác dụng chống đông máu lớn hơn và tăng nguy cơ chảy máu. [Lưu ý: Các chất ức chế CYP quan trọng nhất là erythromycin, ketoconazole và ritonavir, vì mỗi chất ức chế một số isozyme CYP.]
b. Phản ứng pha I không liên quan đến hệ thống P450
Bao gồm quá trình oxy hóa amine (ví dụ, quá trình oxy hóa catecholamine hoặc histamine), quá trình khử hydrogen của rượu (ví dụ, quá trình oxy hóa ethanol), quá trình esterases (ví dụ, quá trình chuyển hóa aspirin trong gan) và thủy phân (ví dụ, procaine).
2. Pha II
Pha này bao gồm các phản ứng liên hợp. Nếu chất chuyển hóa từ pha I đủ phân cực, nó có thể được bài tiết qua thận. Tuy nhiên, nhiều chất chuyển hóa pha I vẫn còn quá ưa béo để có thể được bài tiết. Phản ứng liên hợp tiếp theo với chất nền nội sinh, chẳng hạn như axit glucuronic, axit sulfuric, axit axetic hoặc axit amin, tạo ra các hợp chất phân cực, thường hòa tan trong nước nhiều hơn, thường không có hoạt tính điều trị. Một ngoại lệ đáng chú ý là morphine-6-glucuronide, mạnh hơn morphine. Glucuronide hóa là phản ứng liên hợp phổ biến nhất và quan trọng nhất. [Lưu ý: Thuốc đã có nhóm –OH, –NH2 hoặc –COOH có thể đi vào pha II trực tiếp và liên hợp mà không cần chuyển hóa pha I trước đó (Hình 3).] Các sự liên hợp của thuốc có độ phân cực cao sau đó được bài tiết qua thận hoặc mật.
Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/profile.php?id=61550892771585
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/duoc-dong-hoc-pharmacokinetics-phan-2/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!







{kind=link}