





























III. Sự hấp thu thuốc
B. Các yếu tố ảnh hưởng đến sự hấp thu thuốc
1. Ảnh hưởng của pH đến sự hấp thu thuốc
Hầu hết các loại thuốc đều là axit yếu hoặc bazơ yếu. Thuốc có tính axit (HA) giải phóng một proton (H+), tạo thành anion tích điện (A−):
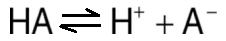
Bazơ yếu (BH+) cũng có thể giải phóng một H+. Tuy nhiên, dạng proton hóa của thuốc bazơ thường tích điện, và việc mất một proton tạo ra bazơ không tích điện (B):
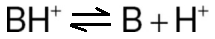
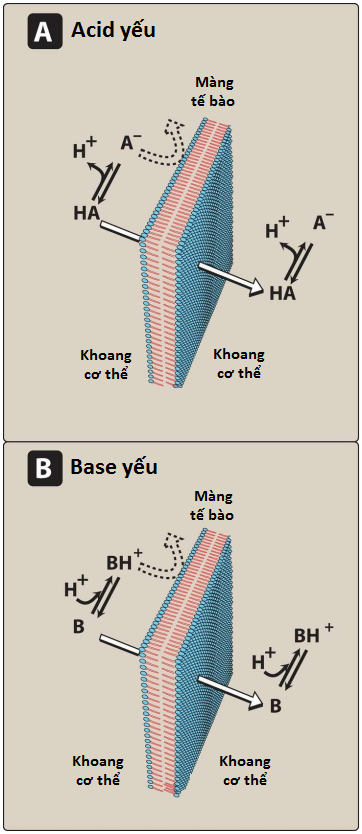
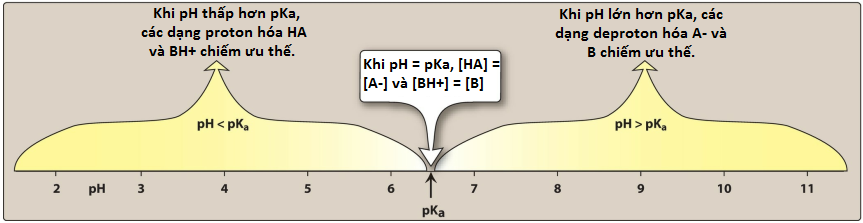
Thuốc đi qua màng dễ dàng hơn nếu nó không tích điện (Hình 1). Do đó, đối với một axit yếu, HA không tích điện, đã proton hóa có thể thấm qua màng, còn A− thì không. Đối với một bazơ yếu, dạng không tích điện B thấm qua màng tế bào, nhưng dạng proton hóa BH+ thì không. Do đó, nồng độ hiệu dụng của dạng thấm của từng loại thuốc tại vị trí hấp thu của nó được xác định bởi các nồng độ tương đối của các dạng tích điện và không tích điện. Tỷ lệ giữa hai dạng này, lần lượt, được xác định bởi độ pH tại vị trí hấp thu và bởi độ mạnh của axit hoặc bazơ yếu, được biểu thị bằng hằng số ion hóa, pKa (Hình 2). [Lưu ý: pKa là thước đo độ mạnh tương tác của hợp chất với proton. pKa của thuốc càng thấp thì tính axit càng cao. Ngược lại, pKa càng cao thì tính bazơ của thuốc càng cao.] Cân bằng phân phối đạt được khi dạng thấm của thuốc đạt được nồng độ bằng nhau trong tất cả các khoang nước của cơ thể.
2. Lưu lượng máu đến vị trí hấp thụ
Ruột nhận được nhiều lưu lượng máu hơn dạ dày, vì vậy sự hấp thụ từ ruột được tạo điều kiện hơn dạ dày. [Lưu ý: Sốc làm giảm nghiêm trọng lưu lượng máu đến các mô da, do đó làm giảm thiểu sự hấp thụ từ việc dùng thuốc dưới da.]
3. Tổng diện tích bề mặt có sẵn để hấp thu
Với bề mặt giàu viền bàn chải chứa vi nhung mao, ruột có diện tích bề mặt gấp khoảng 1000 lần so với dạ dày, giúp hấp thu thuốc qua ruột hiệu quả hơn.
4. Thời gian tiếp xúc tại bề mặt hấp thu
Nếu thuốc di chuyển qua đường tiêu hóa quá nhanh, như có thể xảy ra khi bị tiêu chảy nặng, thì thuốc sẽ không được hấp thụ tốt. Ngược lại, bất cứ điều gì làm chậm quá trình vận chuyển thuốc từ dạ dày đến ruột đều làm chậm tốc độ hấp thụ. [Lưu ý: Sự hiện diện của thức ăn trong dạ dày vừa làm loãng thuốc vừa làm chậm quá trình làm rỗng dạ dày. Do đó, thuốc uống cùng bữa ăn thường được hấp thu chậm hơn.]
5. Độ biểu hiện của P-glycoprotein
P-glycoprotein là một protein vận chuyển xuyên màng có trách nhiệm vận chuyển nhiều phân tử khác nhau, bao gồm thuốc, qua màng tế bào (Hình 3). Nó được biểu hiện trong các mô trên khắp cơ thể, bao gồm gan, thận, nhau thai, ruột và mao mạch não, và tham gia vào quá trình vận chuyển thuốc từ mô vào máu. Nghĩa là, nó “bơm” thuốc ra khỏi tế bào. Do đó, ở những vùng có độ biểu hiện cao, P-glycoprotein làm giảm quá trình hấp thu thuốc. Ngoài việc vận chuyển nhiều loại thuốc ra khỏi tế bào, nó còn liên quan đến tình trạng kháng đa thuốc.

C. Sinh khả dụng
Sinh khả dụng là tốc độ và mức độ mà một loại thuốc được dùng đến được tuần hoàn hệ thống. Ví dụ, nếu 100 mg thuốc được dùng qua đường uống và 70 mg được hấp thu không đổi, thì sinh khả dụng là 0.7 hoặc 70%. Việc xác định sinh khả dụng rất quan trọng để tính liều lượng thuốc cho các đường dùng không phải đường tĩnh mạch.
1. Xác định sinh khả dụng
Sinh khả dụng được xác định bằng cách so sánh nồng độ thuốc trong huyết tương sau một đường dùng cụ thể (ví dụ, dùng qua đường uống) với nồng độ đạt được khi dùng qua đường tĩnh mạch. Sau khi dùng qua đường tĩnh mạch, 100% thuốc nhanh chóng đi vào tuần hoàn. Khi dùng thuốc qua đường uống, chỉ một phần liều dùng xuất hiện trong huyết tương. Bằng cách biểu diễn nồng độ thuốc trong huyết tương theo thời gian, có thể đo được diện tích dưới đường cong (AUC). Sơ đồ mô tả quá trình xác định sinh khả dụng được cung cấp trong Hình 4.
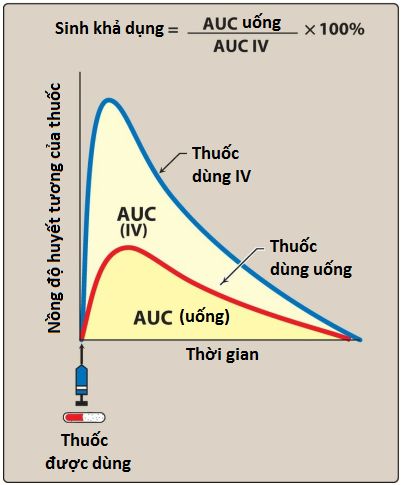
2. Các yếu tố ảnh hưởng đến sinh khả dụng
Ngược lại với đường tiêm tĩnh mạch, mang lại sinh khả dụng 100%, thuốc dùng đường uống thường trải qua quá trình chuyển hóa lần đầu. Sự chuyển hóa sinh học này, cùng với các đặc tính hóa học và vật lý của thuốc, quyết định tốc độ và mức độ mà thuốc đến được tuần hoàn hệ thống.
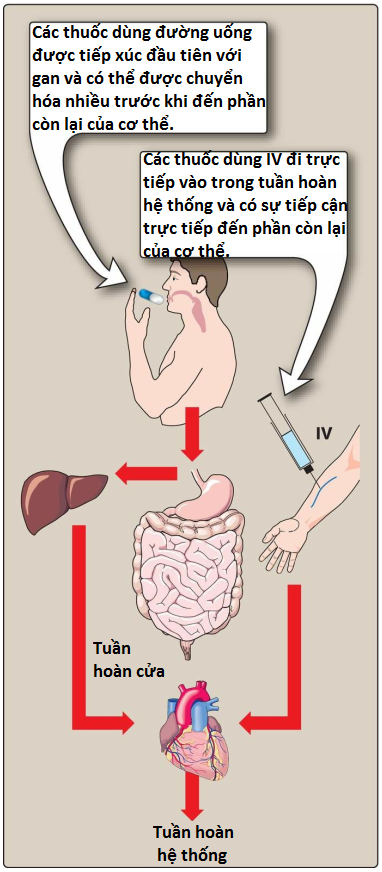
a. Chuyển hóa lần đầu qua gan
Khi một loại thuốc được hấp thu từ đường tiêu hóa, thuốc sẽ đi vào tuần hoàn cửa trước khi vào tuần hoàn toàn thân (Hình 5). Nếu thuốc được chuyển hóa nhanh chóng ở gan hoặc thành ruột trong quá trình chuyển hóa ban đầu này, lượng thuốc không đổi đi vào tuần hoàn hệ thống sẽ giảm. Đây được gọi là chuyển hóa lần đầu qua gan. [Lưu ý: Chuyển hóa lần đầu qua ruột hoặc gan hạn chế hiệu quả của nhiều loại thuốc uống. Ví dụ, hơn 90% nitroglycerin được đào thải trong quá trình chuyển hóa lần đầu qua gan. Do đó, thuốc chủ yếu được dùng qua đường ngậm dưới lưỡi, qua da hoặc tiêm tĩnh mạch.] Các loại thuốc có quá trình chuyển hóa lần đầu qua gan cao nên được dùng với liều lượng đủ để đảm bảo đủ lượng thuốc có hoạt tính đến được vị trí tác dụng mong muốn.
b. Độ hòa tan của thuốc
Thuốc rất ưa nước được hấp thu kém vì không thể đi qua màng tế bào giàu lipid. Nghịch lý thay, thuốc cực kỳ ưa mỡ cũng được hấp thu kém vì chúng không hòa tan trong dịch cơ thể và do đó không thể tiếp cận bề mặt tế bào. Để thuốc được hấp thu dễ dàng, thuốc phải phần lớn là ưa mỡ, nhưng vẫn có một số độ hòa tan trong dung dịch nước. Đây là một lý do tại sao nhiều loại thuốc là axit yếu hoặc bazơ yếu.
c. Không ổn định về mặt hóa học
Một số loại thuốc, chẳng hạn như penicillin G, không ổn định ở độ pH của dịch dạ dày. Một số loại khác, chẳng hạn như insulin, bị phá hủy trong đường tiêu hóa bởi các enzym phân hủy.
d. Bản chất của công thức thuốc
Sự hấp thu thuốc có thể bị thay đổi bởi các yếu tố không liên quan đến hóa học của thuốc. Ví dụ, kích thước hạt, dạng muối, đa hình tinh thể, bao tan trong ruột và sự hiện diện của các tá dược (như chất kết dính và chất phân tán) có thể ảnh hưởng đến khả năng hòa tan và do đó làm thay đổi tốc độ hấp thu.
D. Tương đương sinh học và các loại tương đương khác
Hai công thức thuốc được coi là tương đương sinh học nếu chúng cho thấy sinh khả dụng tương đương và thời gian tương tự để đạt nồng độ đỉnh trong máu. Hai công thức thuốc được coi là tương đương điều trị nếu chúng tương đương bào chế (tức là chúng có cùng dạng bào chế, chứa cùng hoạt chất ở cùng nồng độ và sử dụng cùng đường dùng) với hồ sơ lâm sàng và an toàn tương tự. Do đó, tương đương điều trị đòi hỏi các sản phẩm thuốc phải tương đương sinh học và tương đương về mặt dược phẩm.
IV. Sự phân phối thuốc
Phân phối thuốc là quá trình có thể đảo ngược mà thuốc rời khỏi dòng máu và đi vào dịch ngoại bào và các mô. Đối với thuốc được tiêm tĩnh mạch, sự hấp thu không phải là một yếu tố, và giai đoạn đầu ngay sau khi dùng thuốc là giai đoạn phân phối, trong đó thuốc nhanh chóng rời khỏi tuần hoàn và đi vào các mô (Hình 6). Sự phân phối thuốc từ huyết tương đến mô kẽ phụ thuộc vào cung lượng tim và lưu lượng máu tại chỗ, tính thấm mao mạch, thể tích mô, mức độ liên kết của thuốc với protein huyết tương và mô, và tính ưa mỡ tương đối của thuốc.
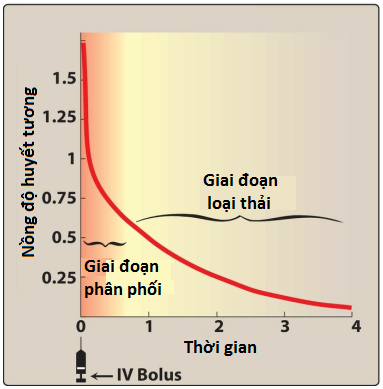
A. Lưu lượng máu
Tốc độ dòng máu chảy đến các mao mạch mô thay đổi rất nhiều. Ví dụ, dòng máu chảy đến “các cơ quan giàu mạch máu” (não, gan và thận) lớn hơn dòng máu chảy đến các cơ xương. Mô mỡ, da và nội tạng có tốc độ dòng máu chảy còn thấp hơn nữa. Sự thay đổi trong dòng máu chảy một phần giải thích thời gian gây mê ngắn do tiêm tĩnh mạch propofol (xem loạt bài viết sau). Lưu lượng máu cao, cùng với tính ưa mỡ cao của propofol, cho phép phân phối nhanh vào CNS và gây mê. Sự phân phối chậm hơn sau đó đến cơ xương và mô mỡ làm giảm nồng độ trong huyết tương để thuốc khuếch tán ra khỏi CNS, theo gradient nồng độ và ý thức được phục hồi.
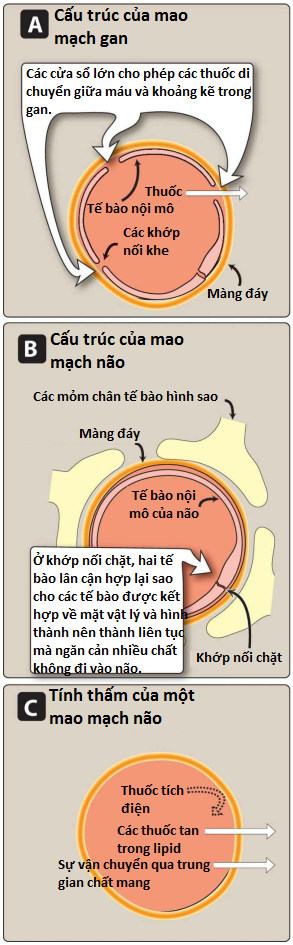
B. Tính thấm mao mạch
Độ thấm mao mạch được xác định bởi cấu trúc mao mạch và bản chất hóa học của thuốc. Cấu trúc mao mạch thay đổi theo tỷ lệ màng đáy được tiếp xúc bởi các khe nối giữa các tế bào nội mô. Ở gan và lách, một phần đáng kể của màng đáy được tiếp xúc do các mao mạch lớn, không liên tục mà các protein huyết tương lớn có thể đi qua (Hình 7A). Ở não, cấu trúc mao mạch là liên tục và không có khe nối (Hình 7B). Để vào não, thuốc phải đi qua các tế bào nội mô của mao mạch CNS hoặc trải qua quá trình vận chuyển tích cực. Ví dụ, một chất vận chuyển cụ thể mang levodopa vào não. Thuốc hòa tan trong lipid dễ dàng thâm nhập vào CNS vì chúng hòa tan trong màng tế bào nội mô. Ngược lại, thuốc ion hóa hoặc phân cực thường không đi vào CNS vì chúng không thể đi qua các tế bào nội mô không có khe nối (Hình 7C). Các tế bào nằm cạnh nhau chặt chẽ này tạo ra các mối nối chặt mà tạo thành hàng rào máu não.
C. Sự liên kết của thuốc với proteins huyết tương và mô
1. Liên kết với protein huyết tương
Liên kết thuận nghịch với protein huyết tương cô lập thuốc ở dạng không khuếch tán và làm chậm quá trình vận chuyển ra khỏi khoang mạch máu. Albumin là protein liên kết thuốc chính và có thể hoạt động như một kho chứa thuốc. Khi nồng độ thuốc tự do giảm do đào thải, thuốc liên kết sẽ tách khỏi albumin. Điều này duy trì nồng độ thuốc tự do dưới một tỷ lệ không đổi của tổng lượng thuốc trong huyết tương.
2. Liên kết với protein mô
Nhiều loại thuốc tích tụ trong mô, dẫn đến nồng độ trong mô cao hơn so với trong dịch kẽ và máu. Thuốc có thể tích tụ do liên kết với lipid, protein hoặc axit nucleic. Thuốc cũng có thể trải qua quá trình vận chuyển tích cực vào mô. Kho chứa mô có thể đóng vai trò là nguồn thuốc chính và kéo dài tác dụng của thuốc hoặc gây độc tính tại chỗ của thuốc. (Ví dụ, acrolein, chất chuyển hóa của cyclophosphamide, có thể gây viêm bàng quang xuất huyết vì nó tích tụ trong bàng quang.)
D. Tính ưa mỡ (lipid)
Bản chất hóa học của thuốc ảnh hưởng mạnh đến khả năng đi qua màng tế bào của thuốc. Thuốc ưa mỡ dễ dàng di chuyển qua hầu hết các màng sinh học. Những loại thuốc này hòa tan trong màng lipid và thâm nhập vào toàn bộ bề mặt tế bào. Yếu tố chính ảnh hưởng đến sự phân bố của thuốc ưa mỡ là lưu lượng máu đến khu vực đó. Ngược lại, thuốc ưa nước không dễ dàng đi qua màng tế bào và phải đi qua các khe nối.
Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/61550892771585/
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/duoc-dong-hoc-pharmacokinetics-phan-1/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!






{kind=link}