





























III. Sự tổ chức của gene globin
Để hiểu được các bệnh do các thay đổi về mặt di truyền trong cấu trúc hay sự tổng hợp của hemoglobin thì cần phải nắm rõ cách mà các genes hemoglobin (giúp tổng hợp các chuỗi globin khác nhau) được tổ chức về mặt cấu trúc thành các họ gene, và cũng nắm rõ cách chúng biểu hiện. Sự biểu hiện của một gene globin bắt đầu trong các tiền thân của tế bào hồng cầu, nơi mà trình tự DNA mã hóa gene được phiên mã. Hai introns được cắt ra khỏi để nối 3 exons thành mRNA trưởng thành cần cho sự dịch mã.
A. Họ α-gene
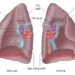
Các gen mã hóa cho các tiểu đơn vị α-globin và β-globin của các chuỗi hemoglobin diễn ra trong 2 cụm gen riêng biệt (hay hai họ gen) nằm trên 2 nhiễm sắc thể khác nhau (Hình 1). Cụm α-gene trên nhiễm sắc thể 16 chứa 2 genes cho các chuỗi α-globin. Cụm này cũng chứa ζ gene được biểu hiện sớm trong quá trình phát triển ở dạng một thành phần tương tự α-globin của hemoglobin thai nhi. (Chú ý: Các họ genes globin cũng chứa các genes giống globin mà không được biểu hiện, nghĩa là thông tin di truyền của nó không được sử dụng để sản xuất các chuỗi globin. Các genes này được gọi là các genes giả (pseudogenes)).

B. Họ β-gene
Cụm β-gene chứa một gen đơn cho chuỗi β-globin, nằm trên nhiễm sắc thể 11 (xem Hình 1). Có thêm 4 genes tương tự β-globin bên trong cụm gene: ε gene (giống với ζ gene, được biểu hiện sớm trong sự phát triển của thai nhi), 2 γ genes (Gγ và Aγ được biểu hiện trong HbF) và δ gene giúp mã hóa cho chuỗi globin được tìm thấy trong hemoglobin thiểu số ở người trưởng thành, HbA2.
IV. Các bệnh hemoglobin
Các bệnh hemoglobin được định nghĩa là một nhóm các rối loạn di truyền được tạo ra bởi sự sản xuất của một phân tử hemoglobin bất thường về mặt cấu trúc, sự tổng hợp các lượng hemoglobin bình thường không đầy đủ, hoặc trường hợp hiếm là khi cả hai điều trên cùng xảy ra. Thiếu máu hồng cầu hình liềm (HbS), bệnh hemoglobin C (HbC), bệnh hemoglobin SC (HbS + HbC = HbSC), và các bệnh thalassemias là các bệnh hemoglobin tiêu biểu mà có thể có các kết cục lâm sàng nghiêm trọng. 3 tình trạng đầu tiên là do sự sản xuất hemoglobin với một trình tự amino acid bị thay đổi (bệnh liên quan đến chất lượng hemoglobin), ngược lại, các bệnh thalassemias được gây ra bởi sự giảm sản xuất hemoglobin bình thường (bệnh liên quan đến số lượng hemoglobin).
A. Bệnh thiếu máu hồng cầu hình liềm (bệnh hemoglobin S)
Bệnh thiếu máu hồng cầu hình liềm là một rối loạn di truyền gây ra bởi một sự thay thế nucleotide (một đột biến điểm) trong gene β-globin. Sự thay đổi trong trình tự amino acid của HbS làm hình dạng của tế bào hồng cầu trở thành hình liềm hay hình trăng khuyết, chứ không phải là hình tròn lõm hai mặt như của một tế bào hồng cầu bình thường xuất hiện trong HbA bình thường. Hình dạng tế bào bất thường này được gọi là sự phát sinh tế bào hồng cầu hình liềm. Thiếu máu hồng cầu hình liềm là rối loạn máu di truyền thường gặp nhất ở Hoa Kỳ, làm ảnh hưởng đến 50,000 người Mỹ. Nó xảy ra chủ yếu ở người Mỹ gốc Phi, tác động đến 1 trong mỗi 500 người Mỹ gốc Phi. Bệnh thiếu máu hồng cầu hình liềm là một rối loạn di truyền theo kiểu lặn trên nhiễm sắc thể thường. Nó xảy ra ở những người mà có 2 alleles đột biến di truyền (một allele của bố và một allele mẹ) giúp mã hóa cho sự tổng hợp các chuỗi β của các phân tử globin. (Chú ý: Chuỗi β-globin đột biến thì được ký hiệu là βS và hemoglobin tạo ra, α2βS 2, sẽ được gọi là HbS). Một đứa trẻ sơ sinh không bắt đầu cho thấy các triệu chứng của bệnh cho đến khi đủ lượng HbF được thay thế bởi HbS để cho sự phát sinh tế bào hồng cầu hình liềm có thể diễn ra. Thiếu máu hồng cầu hình liềm được đặc trưng bởi nhiều giai đoạn đau trong suốt đời cuộc đời (“các cơn”), thiếu máu tan huyết mạn tính với sự tăng bilirubin máu kèm theo và việc dễ mắc các nhiễm khuẩn hơn, thường bắt đầu ở thời thơ ấu. (Chú ý: Đời sống của tế bào hồng cầu trong bệnh hồng cầu hình liềm là dưới 20 ngày, so với 120 ngày của các tế bào hồng cầu bình thường, do đó, gây ra thiếu máu). Các triệu chứng khác bao gồm hội chứng ngực cấp, đột quỵ, rối loạn chức năng lách và thận, và các thay đổi trong xương do tăng sinh tủy. Tuổi thọ trung bình giảm (tuổi trung bình là giữa 40 và 50). Dạng dị hợp tử, biểu hiện ở 1 trong 12 người Mỹ gốc Phi, có một allele bình thường và một allele hồng cầu hình liềm. Các tế bào máu của những dị hợp tử như thế chứa cả HbS và HbA, và những người này có bệnh hồng cầu hình liềm nhẹ. Họ thường không có các dấu hiệu và triệu chứng lâm sàng (nhưng có thể xuất hiện các triệu chứng dưới những điều kiện gắng sức mạnh với mất nước) và có thể có một đời sống bình thường.
1. Sự thay thế amino acid trong các chuỗi β của HbS: Ở một bệnh nhân mắc bệnh thiếu máu hồng cầu hình liềm thì một phân tử HbS sẽ chứa 2 chuỗi α-globin bình thường và 2 chuỗi β-globin đột biến (βS), trong đó glutamate ở vị trí 6 được thay thế bằng valine (Hình 2). Kết quả của sự thay thế các gốc glutamate phân cực tích điện âm thành các gốc valine không phân cực trung tính trong 2 chuỗi β làm cho HbS ít tích điện âm hơn HbA. Vì thế, trong suốt quá trình điện di ở pH kiềm, HbS sẽ di chuyển một cách chậm chạp hơn về phía anode (điện cực dương) so với HbA (Hình 3). Sự điện di hemoglobin thu được từ các tế bào hồng cầu tan thường được sử dụng trong chẩn đoán bệnh thiếu máu hồng cầu hình liềm. Phân tích DNA cũng được sử dụng để chẩn đoán thiếu máu hồng cầu hình liềm.


2. Sự phát sinh tế bào hồng cầu hình liềm và sự thiếu oxy mô: Sự thay thế glutamate tích điện bằng các dạng valine không phân cực sẽ hình thành nên một chỗ lồi kỵ nước trên chuỗi β mà sẽ khớp bổ sung với một vị trí kỵ nước trên chuỗi β của một phân tử HbS khác trong tế bào (Hình 4). Ở cường độ oxygen thấp, HbS không oxygen hóa sẽ polimer hóa bên trong các tế bào hồng cầu, hình thành nên một mạng lưới các polymers dạng sợi không tan làm cứng và méo mó tế bào, tạo ra các tế bào hồng cầu hình liềm vững chắc. Những tế bào hình liềm như vậy thường sẽ làm tắc dòng màu trong các mao mạch hẹp. Sự gián đoạn trong cung cấp oxygen này sẽ dẫn đến thiếu oxygen cục bộ trong mô, gây ra đau và cuối cùng là sự chết do thiếu máu (nhồi máu) của các tế bào lân cận vùng bị tắc. Sự thiếu oxy cũng dẫn đến một sự tăng lên trong số lượng HbS không được oxygen hóa (Chú ý: Đường kính trung bình của các tế bào hồng cầu là 7.5 micrometers, ngược lại đường kính của các vi mạch máu là 3 đến 4 micrometers. So với các tế bào hồng cầu bình thường, các tế bào hình liềm có sự giảm khả năng biến dạng và tăng khả năng dính vào các thành mạch. Điều này làm cho sự di chuyển qua mạch máu nhỏ khó khăn hơn, bằng cách đó, gây ra sự tắc vi mạch máu).

3. Các biến đổi làm tăng sự phát sinh tế bào hồng cầu hình liềm: Mức độ của sự phát sinh tế bào hồng cầu hình liềm và vì thế, độ nghiêm trọng của bệnh được tăng cường bởi bất cứ sự biến đổi nào mà làm tăng lượng HbS ở dạng deoxy (nghĩa là làm giảm ái tính với oxygen của HbS). Những biến đổi này bao gồm giảm pO2, tăng pCO2, giảm pH, mất nước và tăng nồng độ của 2,3-BPG trong các tế bào hồng cầu.
4. Điều trị: Liệu pháp điều trị liên quan đến sự cấp nước đầy đủ, giảm đau, liệu pháp kháng sinh tăng cường nếu như nhiễm khuẩn xuất hiện và truyền máu ở những bệnh nhân có nguy cơ tử vong cao do tắc các mạch máu. Truyền máu gián đoạn bằng các tế bào hồng cầu khối làm giảm nguy cơ đột quỵ, nhưng các lợi ích phải được cân nhắc so với biến chứng của truyền máu, bao gồm quá tải sắt có thể gây ra chứng nhiễm hemosiderin, các nhiễm khuẩn do truyền máu và các biến chứng miễn dịch. Hydroxyurea (hydroxycarbamide), một thuốc chống khối u, thì hữu ích về mặt liệu pháp điều trị bởi vì nó làm tăng các mức HbF tuần hoàn, làm giảm sự hình thành của tế bào hồng cầu hình liềm. Điều này dẫn đến giảm tần suất các cơn đau và giảm tỷ lệ tử vong. Cấy ghép tế bào gốc có thể được thực hiện (Chú ý: Mức độ bệnh tật và tỷ lệ tử vong liên quan đến bệnh thiếu máu hồng cầu hình liềm đã đưa đến việc bao gồm các xét nghiệm theo dõi bệnh lúc mới sinh để cho phép liệu pháp kháng sinh dự phòng được bắt đầu sớm sau sinh với đứa trẻ mắc bệnh).
5. Lợi thế chọn lọc có thể có của trạng thái dị hợp tử: Tần số cao của đột biến βS ở những người châu Phi da đen, mặc cho các tác động gây tổn thương của nó ở trạng thái dị hợp tử, cho thấy rằng có một lợi thế có chọn lọc tồn tại đối với những người mang dị hợp tử. Ví dụ, các dị hợp tử đối với gene hồng cầu hình liềm thì ít mắc sốt rét nghiêm trọng gây ra bởi ký sinh trùng Plasmodium falciparum. Vi sinh vật này dành một phần bắt buộc của chu kỳ sống trong các tế bào hồng cầu. Một giả thuyết đưa ra là bởi vì những tế bào hồng cầu ở những người dị hợp tử đối với HbS có một đời sống ngắn hơn bình thường, nên các ký sinh trùng không thể hoàn thành giai đoạn nội bào trong sự phát triển của nó. Điều này có thể cung cấp một lợi thế chọn lọc đối với các thể dị hợp tử sống ở những vùng mà sốt rét là một nguyên nhân chính gây tử vong. Ví dụ, ở châu Phi, sự phân bố của bệnh thiếu máu hồng cầu hình liềm thì tương tự với sự phân bố của bệnh sốt rét.
B. Bệnh hemoglobin C
Giống như HbS, HbC là một biến thể hemoglobin mà có một sự thay thế amino acid đơn ở vị trí thứ 6 của chuỗi β-globin (xem Hình 2). Tuy nhiên, trong HbC, một lysine được thay thế cho glutamate (so với một sự thay thế bằng valine trong HbS). (Chú ý: Sự thay thế này làm cho HbC di chuyển chậm hơn về phía anode hơn HbA hay HbS [xem Hình 3]). Các bệnh nhân hiếm đồng hợp tử đối với HbC nhìn chung là mắc thiếu máu tán huyết mạn tính tương đối nhẹ. Họ không trải qua các cơn nhồi máu và không bắt buộc thực hiện liệu pháp điều trị đặc hiệu nào.
C. Bệnh hemoglobin SC
Bệnh HbSC là một bệnh trong số các bệnh tạo tế bào hồng cầu hình liềm khác. Trong bệnh này, một số chuỗi β-globin có đột biến hồng cầu hình liềm, ngược lại các chuỗi β-globin khác mang đột biến được tìm thấy trong bệnh HbC. (Chú ý: Các bệnh nhân mắc HbSC thì ở trạng thái dị hợp tử đôi). Chúng được gọi là các dị hợp tử hỗn hợp bởi vì cả hai genes β-globin của chúng đều bất thường, mặc dù là các bất thường khác nhau). Mức hemoglobin có xu hướng cao hơn trong bệnh HbSC so với bệnh thiếu máu hồng cầu hình liềm và thậm chí có thể ở giới hạn dưới của khoảng bình thường. Lâm sàng của những người trưởng thành mắc thiếu máu HbSC khác so với bệnh thiếu máu hồng cầu hình liềm, trong đó, các triệu chứng như các cơn đau thì ít thường xuyên và ít nghiêm trọng hơn. Tuy nhiên, có sự biến đổi đáng kể trên lâm sàng.
D. Bệnh methemoglobin
Sự oxy hóa của sắt trong heme của hemoglobin từ Fe2+ thành Fe3+ tạo ra methemoglobin, không thể kết hợp với O2. Sự oxy hóa này có thể mắc phải và được gây ra bởi tác động của một số thuốc nhất định, như nitrates hay các sản phẩm nội sinh như các gốc oxygen phản ứng. Sự oxy hóa cũng có thể do các khiếm khuyết bẩm sinh, ví dụ như, một sự thiếu hụt NADH-cytochrome b5 reductase (còn được gọi là NADH-methemoglobin reductase), là enzyme chịu trách nhiệm cho sự chuyển đổi của methemoglobin (Fe3+) thành hemoglobin (Fe2+), dẫn đến sự tích tụ của methemoglobin (Hình 5). (Chú ý: Các tế bào hồng cầu của trẻ mới sinh có gần một nửa khả năng so với người lớn trong việc làm giảm methemoglobin). Ngoài ra, các đột biến hiếm trong chuỗi α-globin và β-globin có thể làm sản xuất ra HbM, một hemoglobin bất thường kháng lại reductase. Bệnh methemoglobin được đặc trưng bởi màu da “xanh tím ngả nâu” (chocolate cyanosis) (một màu xanh của da và niêm mạc và máu có màu nâu) là do màu tối của methemoglobin. Các triệu chứng thì liên quan đến mức độ thiếu oxy mô và bao gồm mệt mỏi, đau đầu và khó thở. Trong các trường hợp hiếm, hôn mê và tử vong có thể xảy ra. Điều trị được tiến hành với xanh methylene, chất được oxy hóa bởi Fe3+ và Fe3+ bị khử thành Fe2+.

E. Bệnh thalassemia
Các bệnh thalassemias là các bệnh tán huyết di truyền mà trong đó sự mất cân bằng xảy ra trong quá trình tổng hợp các chuỗi globin. Nhóm bệnh này là các rối loạn đơn gene thường gặp nhất ở người. Bình thường, sự tổng hợp các chuỗi α-globin và β-globin thì được điều phối để cho mỗi chuỗi α-globin sẽ có một chuỗi β-globin tương ứng. Điều này dẫn đến sự hình thành của α2β2 (HbA). Trong các bệnh thalassemias, sự tổng hợp của chuỗi α-globin hoặc β-globin bị khiếm khuyết và nồng độ hemoglobin bị giảm. Một bệnh thalassemia có thể được gây ra bởi nhiều các loại đột biến, bao gồm mất toàn bộ gene hoặc thay thế hoặc mất một trong nhiều nucleotides của DNA. (Chú ý: Mỗi bệnh thalassemia có thể được phân loại là một rối loạn mà trong đó không có chuỗi globin nào được sản xuất (α0– hoặc β0-thalassemia), hoặc một rối loạn mà trong đó một số chuỗi được tổng hợp nhưng ở mức độ giảm đi [α+– hoặc β+-thalassemia]).
1. Các β-thalassemias: Trong những rối loạn này, sự tổng hợp các chuỗi β-globin bị giảm hoặc không có, thường là kết quả của các đột biến điểm làm ảnh hưởng đến sự sản xuất của mRNA chức năng. Tuy nhiên, sự tổng hợp chuỗi α-globin thì bình thường. Các chuỗi α-globin thừa thì không thể hình thành nên các tetramers ổn định và do đó nó lắng đọng lại, làm chết sớm các tế bào mà ban đầu có thể trở thành các tế bào hồng cầu trưởng thành. Sự tăng trong α2δ2 (HbA2) và α2γ2 (HbF) cũng diễn ra. Chỉ có 2 bản sao của gene β-globin trong mỗi tế bào (mỗi gene trên một nhiễm sắc thể số 11). Vì thế, những người mà có các khiếm khuyết gene β-globin thì sẽ mắc β-thalassemia nhẹ nếu như họ chỉ có một gene β-globin khiếm khuyết hoặc β-thalassemia nặng (thiếu máu Cooley) nếu như cả 2 genes đều bị khiếm khuyết (Hình 6). Bởi vì gene β-globin thì không được biểu hiện cho đến cuối quá trình phát triển trước khi sinh nên các biểu hiện thực thể của các bệnh β-thalassemia chỉ xuất hiện vài tháng sau khi sinh. Những người mắc β-thalassemia nhẹ thường sản xuất được một số chuỗi β và thường không cần điều trị đặc hiệu. Tuy nhiên, các đứa trẻ sinh ra với β-thalassemia nặng thì dường như khỏe mạnh lúc sinh nhưng trở nên thiếu máu nghiêm trọng do sự sản xuất hồng cầu không hiệu quả, thường là trong suốt năm đầu tiên hoặc năm thứ hai của cuộc đời. Các sự thay đổi về xương là kết quả của sự tạo máu ngoài tủy xương cũng được quan sát thấy. Những bệnh nhân này yêu cầu sự truyền máu thường xuyên. (Chú ý: Mặc dù sự điều trị này thì có thể cứu được mạng sống nhưng tác động tích lũy của truyền máu là quá tải sắt. Việc sử dụng liệu pháp chelat hóa sắt sẽ giúp cải thiện trạng thái bệnh tật và tỷ lệ tử vong). Phương án cứu chữa duy nhất hiện có là cấy ghép tế bào gốc tạo máu.

2. Các α-thalassemias: Trong các rối loạn này, sự tổng hợp của các chuỗi α-globin bị giảm hoặc không có, thường là do các đột biến mất vật liệu di truyền. Bởi vì mỗi bộ gen của con người chứa 4 bản sao của gen α-globin (2 trên mỗi nhiễm sắc thể 16), nên có nhiều mức độ của suy giảm chuỗi α-globin (Hình 7). Nếu như một trong 4 alleles mã hóa cho một protein globin khiếm khuyết thì người đó sẽ được gọi là “thể ẩn” của α-thalassemia bởi vì không có các biểu hiện thực thể của bệnh. Nếu như 2 alleles α-globin mã hóa cho các proteins globin khiếm khuyết thì người bệnh sẽ mắc bệnh thalassemia nhẹ. Nếu như 3 alleles α-globin mã hóa cho các proteins globin khiếm khuyết thì cá thể sẽ mắc bệnh hemoglobin H (β4), một thiếu máu tán huyết với mức độ nặng thay đổi. Nếu như cả 4 alleles mã hóa cho các proteins globin đều bị khiếm khuyết thì sẽ mắc bệnh hemoglobin Bart (γ4), gây ra phù thai và chết thai bởi vì các chuỗi α-globin thì cần cho sự tổng hợp của HbF (Chú ý: Các ưu thế của dị đồng tử chống lại bệnh sốt rét thì được phát hiện thấy trong cả bệnh α- và β-thalassemias).
V. Tóm tắt chương
- Hemoglobin A (HbA), hemoglobin chính ở người trường thành, được tạo thành từ 4 chuỗi polypeptide (2 chuỗi α và 2 chuỗi β, α2β2) kết hợp lại với nhau bởi các tương tác không phải cộng hóa trị (Hình 8).

- Các tiểu đơn vị chiếm lấy các vị trí tương đối khác nhau trong deoxyhemoglobin so với oxyhemoglobin. Dạng deoxy của Hb được gọi là cấu hình “T”, hay taut (căng thẳng). Nó có một cấu trúc ràng buộc làm giới hạn sự vận động của các chuỗi polypeptide. Dạng T là dạng có ái tính thấp với oxygen của Hb.
- Sự liên kết của oxygen (O2) với sắt của heme làm gây ra sự đứt gãy của một số liên kết ion và hydrogen và sự chuyển động của các dimers. Điều này đưa đến sự hình thành của một cấu hình được gọi là “R”, hay cấu hình thư giãn. Dạng R là dạng có ái tính cao với oxygen của Hb.
- Đường cong phân ly oxygen đối với Hb thì có dạng sigmoid (ngược lại với myoglobin, có dạng hyperbol), cho thấy rằng các tiểu đơn vị phối hợp với nhau trong việc liên kết với oxygen. Sự liên kết của một phân tử oxygen ở một nhóm heme làm tăng ái tính với oxygen của các nhóm còn lại trong cùng phân tử Hb (tính phối hợp).
- Khả năng của Hb kết hợp với oxygen thì bị ảnh hưởng thuận nghịch bởi áp lực riêng phần của oxygen (pO2), pH của môi trường, áp lực riêng phần của carbon dioxide (pCO2) và sự có mặt của 2,3-bisphosphoglycerate (2,3-BPG). Ví dụ, sự giải phóng oxygen từ Hb sẽ được tăng cường khi pH hạ xuống hay pCO2 tăng lên (hiệu ứng Bohr), như trong cơ đang luyện tập thể thao và đường cong phân ly oxygen của Hb được dịch chuyển sang bên phải.
- Để ứng phó lâu dài với các tác động của thiếu oxy mô hay thiếu máu mạn tính thì nồng độ 2,3-BPG trong các tế bào hồng cầu sẽ tăng lên. 2,3-BPG liên kết với Hb và làm giảm ái tính với oxygen của nó. Vì thế, nó cũng làm dịch chuyển đường cong phân ly oxygen sang bên phải.
- Hemoglobin thai nhi (HbF) liên kết với 2,3-BPG ít chặt chẽ hơn so với HbA và có một ái tính với oxygen cao hơn.
- Carbon monoxide (CO) liên kết chặt chẽ (nhưng thuận nghịch) với sắt của Hb, hình thành nên carboxyhemoglobin.
- Các bệnh hemoglobin là các rối loạn chủ yếu gây ra bởi sự sản xuất của một phân tử Hb bất thường về mặt cấu trúc như trong thiếu máu tế bào hồng cầu hình liềm hay sự tổng hợp các lượng không đầy đủ của các tiểu đơn vị Hb bình thường như trong các bệnh thalassemias (Hình 9).

Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/profile.php?id=61550892771585
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/cac-proteins-hinh-cau-phan-1/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!






{kind=link}