





























VI. Các rối loạn chuyển hóa amino acids
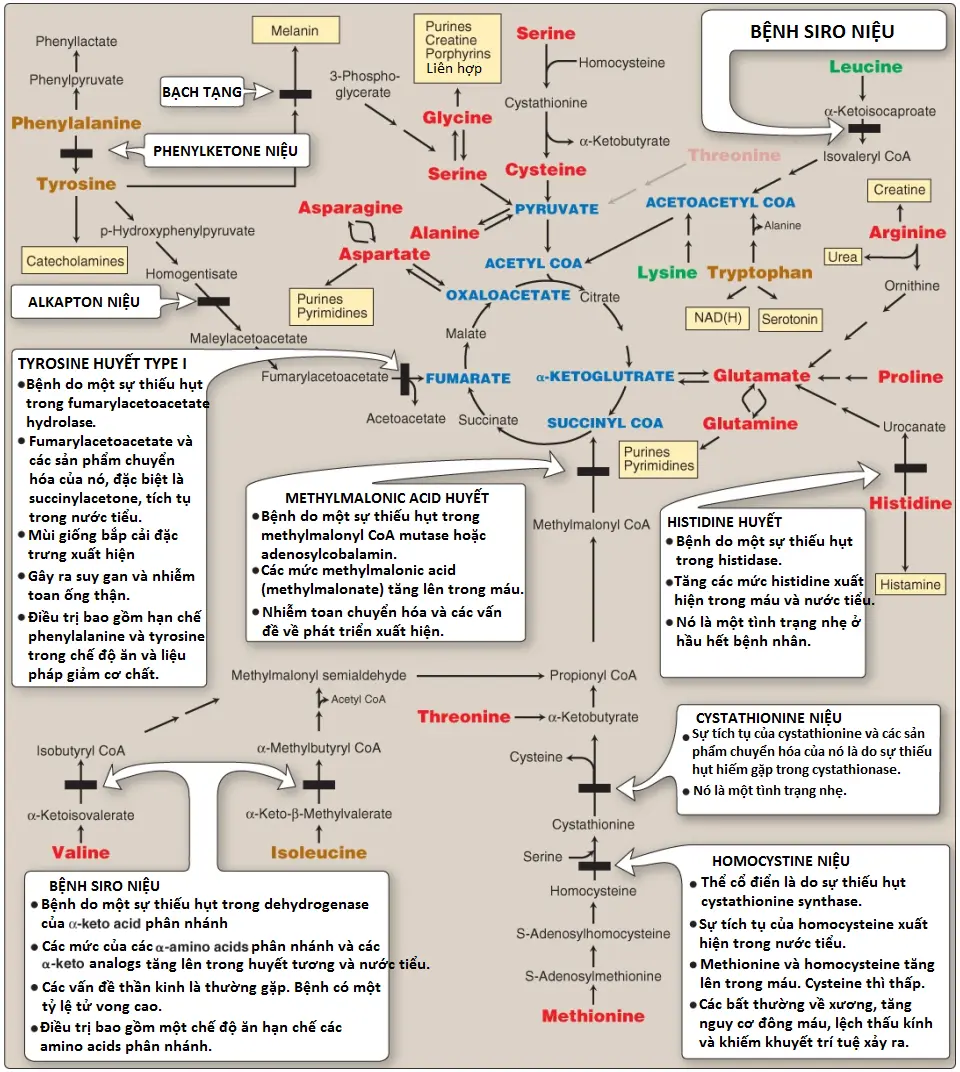
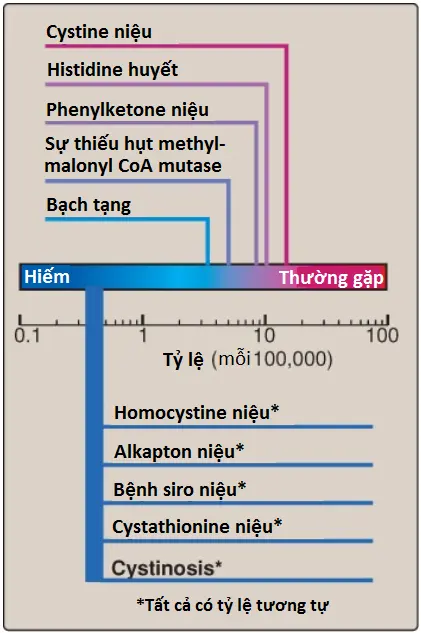
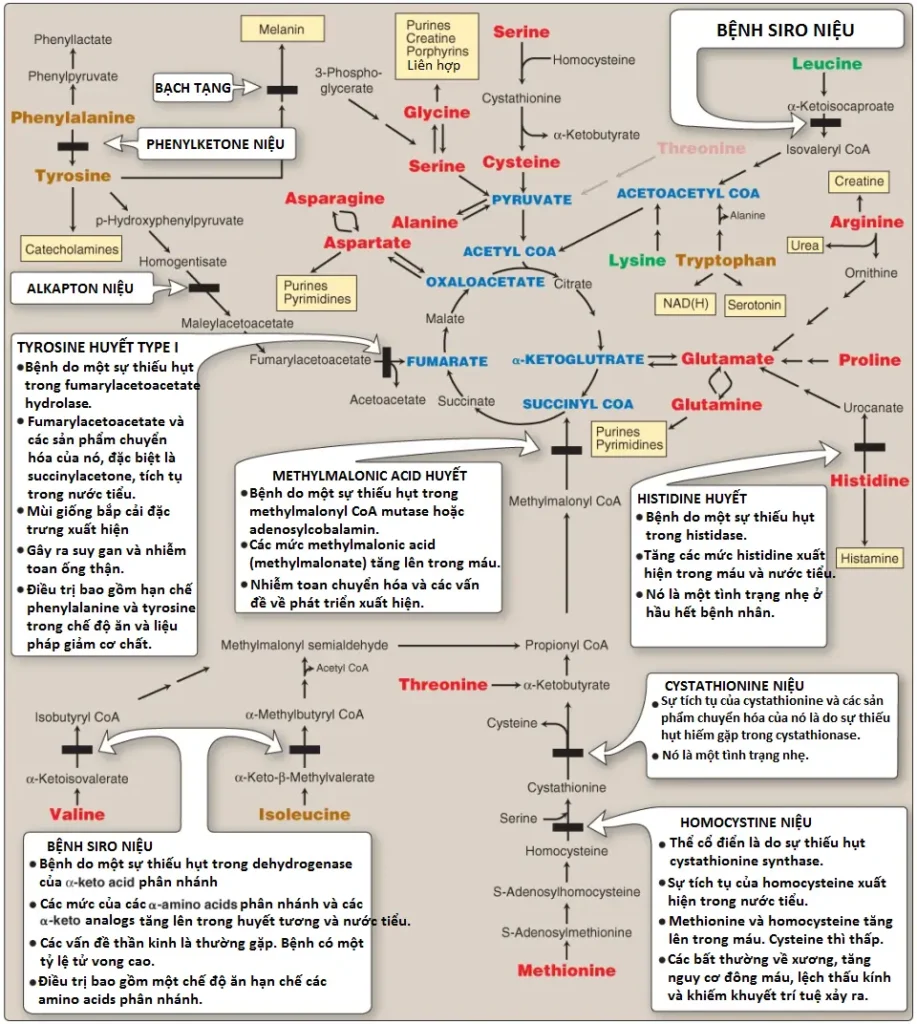
Các rối loạn đơn gen này, một nhóm của các rối loạn chuyển hóa bẩm sinh, nhìn chung là được gây ra bởi các đột biến mất chức năng trong các enzymes liên quan đến sự chuyển hóa amino acid. Các khiếm khuyết di truyền có thể được biểu hiện dưới dạng một sự mất hoạt động enzyme hoàn toàn hay phổ biến hơn là dưới dạng một sự suy giảm trong hoạt động xúc tác. Nếu không điều trị, các rối loạn amino acids hầu như luôn gây ra khuyết tật tật trí tuệ hoặc các bất thường phát triển khác, như là kết quả của sự tích tụ các chất chuyển hóa có hại. Mặc dù có hơn 50 rối loạn đã được mô tả nhưng nhiều trong số đó là hiếm gặp, xảy ra ở <1 trên mỗi 250,000 trong hầu hết các dân số (Hình 14). Tuy nhiên, tựu chung lại thì chúng chiếm một phần rất đáng kể của các bệnh di truyền nhi khoa (Hình 15).
A. Phenylketone niệu
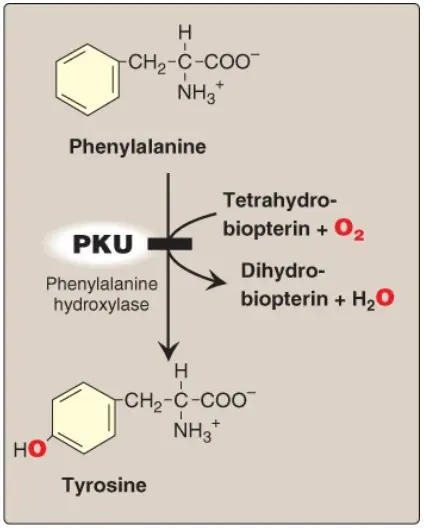
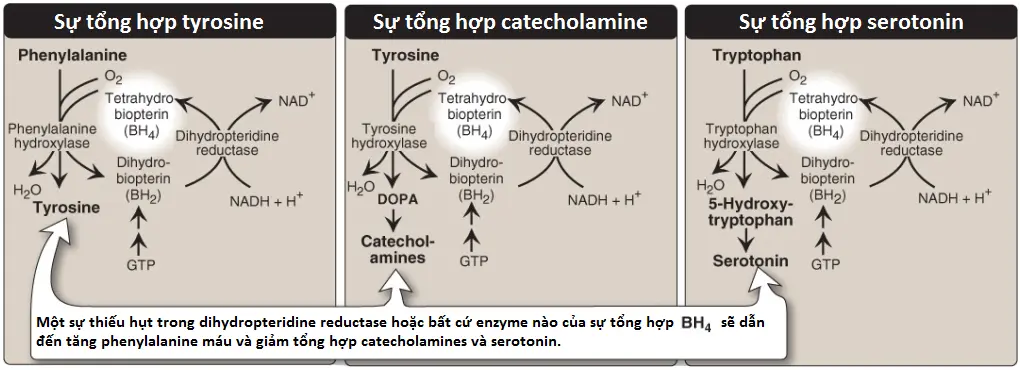
PKU (phenylketonuria) là khiếm khuyết bẩm sinh trong chuyển hóa amino acid thường gặp nhất trên lâm sàng (tỷ lệ 1:15,000). PKU cổ điển là một rối loạn lặn trên nhiễm sắc thể thường do các đột biến mất chức năng trong gen mã hóa cho PAH (Hình 16). Về mặt hóa sinh, PKU được đặc trưng bởi tăng phenylalanine máu. Phenylalaine xuất hiện với các nồng độ cao (10 lần so với bình thường) không chỉ trong huyết tương mà còn trong nước tiểu và các mô cơ thể. Tyrosine, bình thường được hình thành từ phenylalanine bởi PAH, bị suy giảm. Điều trị bao gồm hạn chế phenylalanine trong chế độ ăn và bổ sung tyrosine. (Chú ý: Tăng phenylalanine máu cũng có thể được gây ra bởi các sự thiếu hụt hiếm gặp trong bất cứ enzyme nào trong số một vài enzyme cần cho sự tổng hợp BH4 hoặc trong dihydropteridine reductase, enzyme giúp tái tạo BH4 từ BH2 [Hình 17]. Các sự thiếu hụt như vậy làm tăng một cách gián tiếp các nồng độ phenylalanine, bởi vì PAH cần BH4 như là một coenzyme. BH4 cũng được cần cho tyrosine hydroxylase và tryptophan hydroxylase, là các enzyme xúc tác cho các phản ứng dẫn đến sự hình thành của các chất dẫn truyền kinh kinh, như serotonin và catecholamines. Đơn giản hạn chế phenylalanine trong chế độ ăn thì không thể đảo ngược các tác động lên hệ thống thần kinh trung ương do các sự thiếu hụt trong các chất dẫn truyền thần kinh. Sự bổ sung BH4 và liệu pháp thay thế với L-3,4-dihydroxyphenylalanine [L-DOPA] và 5-hydroxytryptophan [các sản phẩm của các phản ứng tyrosine hydroxylase và tryptophan hydroxylase bị ảnh hưởng] làm cải thiện kết cục lâm sàng trong các biến thể của tăng phenylalanine máu mặc dù đáp ứng là không thể dự đoán được).
1. Các đặc điểm khác: Giống như tên của bệnh, PKU cũng được đặc trưng bởi các mức phenylketone tăng lên trong nước tiểu.
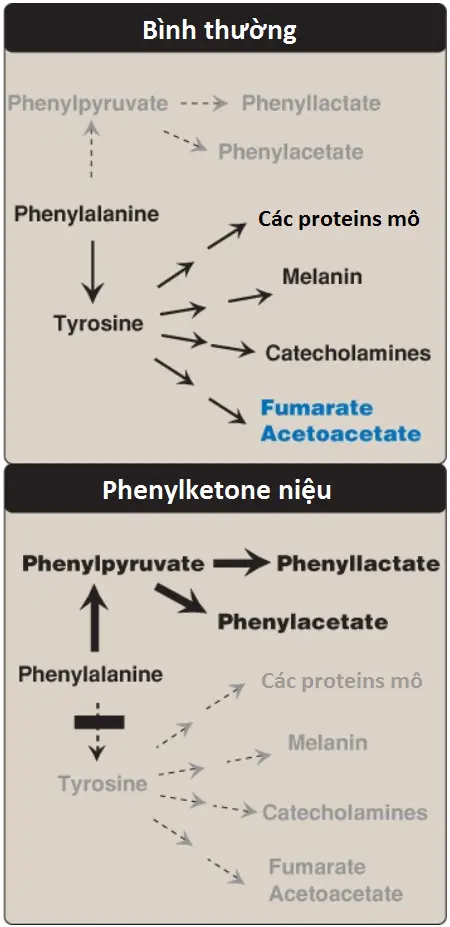
a. Các chất chuyển hóa của phenylalanine tăng lên: Phenylpyruvate (một phenylketone), phenylacetate và phenyllactate, mà không được tạo ra một cách bình thường với các lượng đáng kể trong sự có mặt của PAH chức năng, cũng được tăng lên trong PKU, ngoài phenylalanine (Hình 18). Các chất chuyển hóa này làm cho nước tiểu có mùi mốc.
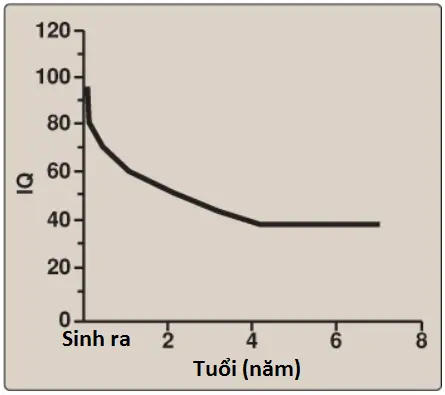
b. Các tác động lên hệ thống thần kinh trung ương: Khuyết tật trí tuệ nghiêm trọng, sự phát triển chậm, tật đầu nhỏ và co giật là các phát hiện đặc trưng trong PKU không điều trị. Người bị ảnh hưởng thường cho thấy các triệu chứng của khuyết tật trí tuệ ở lúc 1 tuổi và hiếm khi có chỉ số IQ trên 50 (Hình 19). (Chú ý: Các biểu hiện lâm sàng này bây giờ hiếm khi được quan sát thấy như là một kết quả của các chương trình theo dõi sơ sinh mà cho phép chẩn đoán và điều trị sớm).
c. Giảm sắc tố: Các bệnh nhân mắc PKU không điều trị có thể cho thấy một sự thiếu hụt sắc tố (tóc nhạt máu, màu da sáng và mắt xanh). Sự hydroxyl hóa của tyrosine bởi tyrosinase cần đồng, là bước đầu tiên trong sự hình thành của sắc tố melanin, bị giảm trong PKU bởi vì tyrosine bị giảm.
2. Theo dõi và chẩn đoán sơ sinh: Chẩn đoán sớm PKU là điều quan trọng bởi vì bệnh thì có thể điều trị được thông qua chế độ ăn uống. Do thiếu các triệu chứng sơ sinh nên xét nghiệm các mức phenylalanine trong máu tăng là cần thiết để phát hiện bệnh. Tuy nhiên, trẻ sơ sinh mắc PKU thường có các mức phenylalanine máu bình thường lúc sinh bởi vì người mẹ làm mất đi mức phenylalanine tăng lên trong máu ở thai nhi bị ảnh hưởng qua nhau thai. Các mức phenylalanine bình thường có thể tồn tại cho đến khi trẻ sơ sinh được cho ăn protein trong 24 đến 48 giờ. Vì thế, các xét nghiệm theo dõi thường được thực hiện sau thời gian này để tránh âm tính giả. Đối với các trẻ mới sinh có kết quả theo dõi dương tính thì chẩn đoán được xác nhận thông qua sự xác định định lượng của các mức phenylalanine.
3. Chẩn đoán trước sinh: PKU cổ điển được gây ra bởi bất cứ đột biến nào trong số 100 hoặc nhiều hơn các đột biến khác nhau trong gen mã hóa PAH. Tần suất của bất cứ đột biến gen nào đều khác giữa các dân số khác nhau và bệnh thường ở dạng dị hợp tử phức đơn (compound heterozygous) (nghĩa là gen PAH có một đột biến khác nhau trong mỗi alen). Mặc cho tính phức tạp này, chẩn đoán trước sinh vẫn có thể thực hiện được.
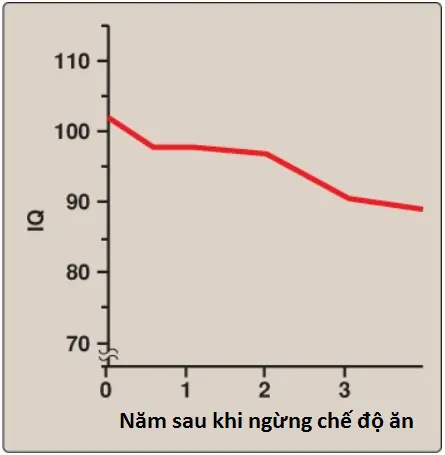
4. Điều trị: Bởi vì hầu hết protein tự nhiên chứa phenylalanine, một amino acid thiết yếu, nên không thể đáp ứng đủ nhu cầu protein của cơ thể mà không vượt quá giới hạn phenylalanine khi tiêu hóa một chế độ ăn bình thường. Vì thế, trong bệnh PKU, mức phenylalanine máu được duy trì gần với khoảng bình thường bằng cách cho ăn các sản phẩm amino acid tổng hợp không có phenylalanine, bổ sung với một số thực phẩm tự nhiên (như trái cây, rau củ và các loại ngũ cốc nhất định) được chọn lọc bởi hàm lượng phenylalanine thấp của chúng. Lượng phenylalanine được điều chỉnh theo mức độ dung nạp của cá nhân khi được đo lường bởi các mức phenylalanine trong máu. Sự điều trị được bắt đầu càng sớm thì tổn thương thần kinh có thể được ngăn chặn càng tốt hơn. Những người mà được điều trị một cách thích hợp có thể có trí tuệ bình thường. (Chú ý: Điều trị phải bắt đầu trong suốt 7 đến 10 ngày đầu tiên của cuộc đời để ngăn cản suy giảm nhận thức). Bởi vì phenylalanine là một amino acid thiết yếu nên việc điều trị quá tích cực mà làm cho các mức phenylalanine máu bên dưới mức bình thường thì cần được tránh. Ở những bệnh nhân mắc PKU, tyrosine không thể được tổng hợp từ phenylalanine và do đó, nó trở thành một amino acid thiết yếu và phải được cung cấp trong chế độ ăn. Sự ngừng chế độ ăn hạn chế phenylalanine ở đầu thời kỳ thơ ấu thì sẽ liên quan đến các kết quả kém trong các kiểm tra IQ. Những bệnh nhân PKU trưởng thành cho thấy sự suy giảm điểm số IQ sau khi ngừng chế độ ăn (Hình 20). Vì thế, hạn chế cả đời phenylalanine trong chế độ ăn thì được khuyến cáo. (Chú ý: Những người mắc PKU được khuyên là tránh aspartame, một chất làm ngọt nhân tạo mà chứa phenylalanine).
5. Phenylketone niệu ở mẹ: Nếu như phụ nữ mắc PKU mà không tuân theo chế độ ăn thấp phenylalanine mang thai, thì con vẫn có thể bị ảnh hưởng bởi hội chứng PKU của người mẹ. Ngay cả khi thai nhi không được di truyền bệnh (nghĩa là thai nhi dị hợp đối với đột biến PAH), phenylalanine trong máu cao ở người mẹ có một tác động gây quái thai, gây ra tật đầu nhỏ và các bất thường tim bẩm sinh ở thai nhi. Bởi vì các đáp ứng phát triển này với mức phenylalanine cao xảy ra trong suốt các tháng đầu tiên của thời kỳ mang thai nên sự kiểm soát chế độ ăn đối với mức phenylalanine trong máu phải được bắt đầu trước khi thụ thai và được duy trì trong suốt thai kỳ.
B. Bệnh siro niệu
MSUD (maple syrup urine disease) là một rối loạn lặn trên nhiễm sắc thể thường hiếm gặp (1:185,000) mà trong đó có một sự thiếu hụt một phần hoặc hoàn toàn BCKD, phức hợp enzyme ty thể mà decarboxyl oxy hóa leucine, isoleucine và valine (xem Hình 11, phần 2). Các BCAAs này và các α-keto acids tương ứng của chúng tích tụ trong máu, gây ra một tác động gây độc mà ảnh hưởng đến các chức năng não bộ. Bệnh được đặc trưng bởi các vấn đề khi cho ăn, nôn mửa, nhiễm toan ketoacid, các thay đổi trong trương lực cơ, các vấn đề thần kinh mà có thể gây ra hôn mê (chủ yếu là do sự tăng lên trong leucine) và một mùi giống siro phong (maple syrup) của nước tiểu do sự tăng lên trong isoleucine. Nếu như không điều trị, bệnh sẽ gây tử vong. Nếu điều trị bị chậm trễ thì khuyết tật trí tuệ sẽ xảy ra.
1. Phân loại: MSUD bao gồm một thể cổ điển và các biến thể khác. Thể cổ điển khởi phát sơ sinh là thể MSUD thường gặp nhất. Các tế bào bạch cầu hoặc các nguyên bào sợi da sinh thiết từ những bệnh nhân này cho thấy ít hoặc không có hoạt động của BCKD. Những đứa trẻ mắc MSUD cổ điển cho thấy các triệu chứng trong một vài ngày đầu tiên. Nếu như không được chẩn đoán và điều trị, MSUD cổ điển sẽ gây tử vong trong vài tuần đầu tiên của cuộc đời. Những bệnh nhân mắc các thể trung bình có mức độ hoạt động của enzyme cao hơn (lên đến 30% so với bình thường). Các triệu chứng thì nhẹ hơn và cho thấy một sự khởi phát từ lúc mới sinh cho đến tuổi thanh thiếu niên. Những bệnh nhân mắc biến thể phụ thuộc thiamine hiếm gặp của MSUD thì đáp ứng với các liều lượng lớn hơn của vitamin này.
2. Theo dõi và chẩn đoán: Như với PKU, chẩn đoán trước sinh và theo dõi sơ sinh thì có sẵn đối với bệnh này và hầu hết những người bị ảnh hưởng là dị hợp tử phức đơn (compound heterozygotes).
3. Điều trị: MSUD được điều trị bởi công thức ăn uống tổng hợp không chứa BCAA, được bổ sung với các lượng giới hạn leucine, isoleucine và valine để cho phép sự tăng trưởng và phát triển bình thường mà không tạo ra các mức gây độc. (Chú ý: Mức leucine tăng lên là nguyên nhân của tổn thương thần kinh trong MSUD và các mức của nó được theo dõi một cách cẩn thận). Chẩn đoán sớm và điều trị thông qua chế độ ăn suốt đời là cần thiết để trẻ mắc MSUD phát triển một cách bình thường. (Chú ý: BCAAs là một nguồn năng lượng quan trọng trong nhiều thời điểm của nhu cầu chuyển hóa và những người mắc MSUD thì có nguy cơ mất bù trong suốt các giai đoạn tăng dị hóa protein).
C. Bạch tạng
Bạch tạng (albinism) đề cập đến một nhóm các tình trạng mà trong đó một sự thiếu hụt trong chuyển hóa tyrosine gây ra một sự thiếu hụt trong sự sản xuất melanin. Các tác động này gây ra sự vắng mặt một phần hoặc hoàn toàn của sắc tố khỏi da, tóc và mắt. Bạch tạng xuất hiện ở nhiều dạng khác nhau và nó có thể được di truyền bởi một trong số các kiểu: lặn trên nhiễm sắc thể thường (kiểu chủ yếu), di truyền trội trên nhiễm sắc thể thường hoặc liên kết với nhiễm sắc thể X. Sự vắng mặt hoàn toàn của sắc tố khỏi tóc, mắt và da (Hình 21), là bệnh bạch tạng mắt da không có tyrosinase (tyrosinase-negative oculocutaneous albinism) (bạch tạng type 1 [type 1 albinism]), do sự vắng mặt hoặc khiếm khuyết trong tyrosinase cần đồng, tyrosinase là enzyme xúc tác cho hai bước đầu tiên trong sự tổng hợp melanin từ tyrosine. Nó là dạng nghiêm trọng nhất của tình trạng này. Ngoài giảm sắc tố, những người bị ảnh hưởng có các suy giảm về thị lực và mắc chứng sợ ánh nắng (ánh nắng mặt trời làm tổn thương mắt của họ). Họ cũng bị tăng nguy cơ mắc ung thư da.
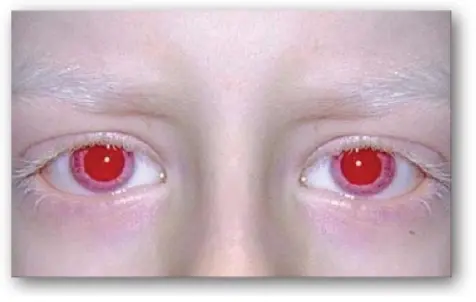
D. Homocysteine niệu
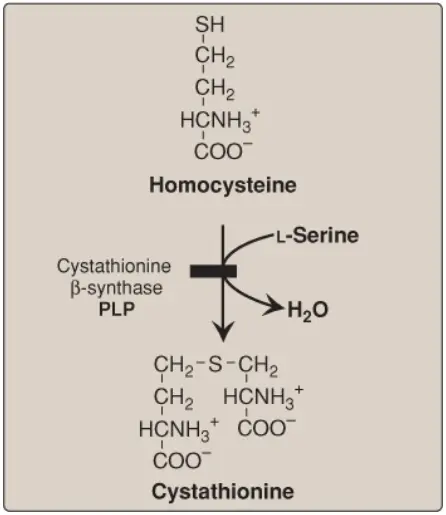
Homocystine niệu (homocystinurias) là một nhóm các rối loạn liên quan đến các khiếm khuyết trong chuyển hóa của Hcy. Các bệnh di truyền lặn trên nhiễm sắc thể thường này được đặc trưng bởi các mức Hcy trong nước tiểu cao, các mức Hcy và methionine trong huyết tương cao và các mức cysteine trong huyết tương thấp. Nguyên nhân thường gặp nhất của homocystine niệu là một khiếm khuyết trong enzyme cystathionine β-synthase, là enzyme chuyển Hcy thành cystathionine (Hình 22). Những người đồng hợp đối với tình trạng thiếu hụt cystathionine β-synthase cho thấy sự lệch thủy tinh thể (ectopia lentis), các bất thường về xương (các chi và các ngón tay dài), khuyết tật trí tuệ và nguy cơ phát triển các cục máu đông tăng lên. Hình thành cục máu đông (thrombosis) là nguyên nhân chính gây tử vong sớm ở những người này. Điều trị bao gồm hạn chế methionine và bổ sung vitamin B12 và folate. Cysteine trở thành một amino acid thiết yếu và phải được bổ sung cho cơ thể. Bởi vì glutathione được tổng hợp từ cysteine (Hình 6, bài viết trước) nên bổ sung cysteine vào trong chế độ ăn cũng hỗ trợ trong việc làm giảm stress oxy hóa. Ngoài ra, một số bệnh nhân cũng đáp ứng với việc sử dụng pyridoxine (vitamin B6) đường uống, là chất được chuyển thành pyridoxal phosphate, coenzyme của cystathionine β-synthase. Những bệnh nhân này thường có một sự khởi phát các triệu chứng lâm sàng nhẹ hơn và trễ hơn so với những bệnh nhân không đáp ứng vitamin B6. (Chú ý: Các sự thiếu hụt trong methylcobalamin [xem Hình 8, phần 1] hoặc N5,N10-MTHF reductase [(MTHFR), xem Hình 12, phần 2] cũng gây ra tăng Hcy).
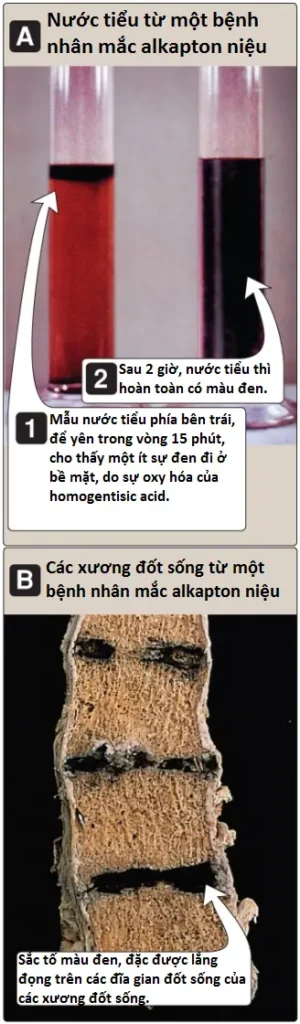
E. Alkapton niệu
Alkapton niệu (alkaptonuria) là một tình trạng acid hữu cơ niệu (organic aciduria) hiếm gặp liên quan đến một sự thiếu hụt trong homogentisic acid oxidase, gây ra sự tích tụ của homogentisic acid (HA), một trung gian trong con đường thoái hóa tyrosine (xem Hình 15, phần 2). Tình trạng này có ba triệu chứng đặc trưng: homogentisic acid niệu (nước tiểu chứa các mức HA tăng lên, là thành phần bị oxy hóa thành một sắc tố sẫm màu khi để yên, như được thể hiện trong Hình 23A), sự khởi phát sớm của viêm khớp trong các khớp lớn và sự lắng đọng của sắc tố đen (ochronosis) trong mô sụn và collagen (xem Hình 23B). Sự nhuộm màu tối của tã lót có thể chỉ điểm bệnh ở trẻ nhỏ, nhưng thường không có triệu chứng xuất hiện cho đến khoảng 40 tuổi. Điều trị bao gồm hạn chế phenylalanine và tyrosine trong chế độ ăn để làm giảm các mức HA. Mặc dù alkapton niệu thì không đe dọa tính mạng nhưng viêm khớp liên quan có thể làm tàn tật nặng. (Chú ý: Các sự thiếu hụt trong fumarylacetoacetate hydrolase, enzyme cuối cùng của sự chuyển hóa tyrosine, gây ra tyrosine huyết type 1 [xem Hình 15, phần 2] và một mùi giống bắp cải đặc trưng của nước tiểu).
F. Methylmalonic acid huyết
Methylmalonic acid huyết (methylmalonic acidemia – MMA) là một rối loạn lặn trên nhiễm sắc thể thường hiếm gặp (1:100,000) được gây ra bởi một sự thiếu hụt trong methylmalonyl CoA mutase, enzyme giúp chuyển L-methylmalonyl CoA thành succinyl CoA. Bởi vì mutase cần vitamin B12 nên bệnh cũng có thể do sự thiếu hụt vitamin B12 nghiêm trọng. Sự thoái hóa của các acids béo có số lẻ carbon, valine, isoleucine, methionine và threonine tất cả đều có thể tạo ra MMA do sự thiếu hụt enzyme này. Sự tăng lên trong methylmalonate máu và nước tiểu có thể gây ra nhiễm toan chuyển hóa. Cũng có thể có một sự tăng lên trong propionyl-CoA, làm tăng cường tình trạng acid niệu với sự tích tụ của thêm propionic acid. Các triệu chứng xuất hiện trong giai đoạn đầu khi trẻ mới sinh, thay đổi tùy theo mức độ thiếu hụt enzyme, bao gồm chậm phát triển, nôn mửa, mất nước, giảm trương lực cơ, co giật, gan to, tăng amoniac máu và một bệnh não tiến triển. Nếu bị nặng mà không điều trị, bệnh có thể dẫn đến khuyết tật trí tuệ, tổn thương gan hoặc thận mạn tính, viêm tụy và hôn mê hoặc tử vong. Điều trị bao gồm một chế độ ăn giàu calo, ít protein với sự bổ sung thêm vitamin B12. Chế độ ăn hạn chế sự ăn vào của isoleucine, threonine, methionine và valine bởi vì các amino acids này có thể dẫn đến sự tích tụ của methylmalonic acid bởi sự thiếu hụt mutase.
VII. Tóm tắt
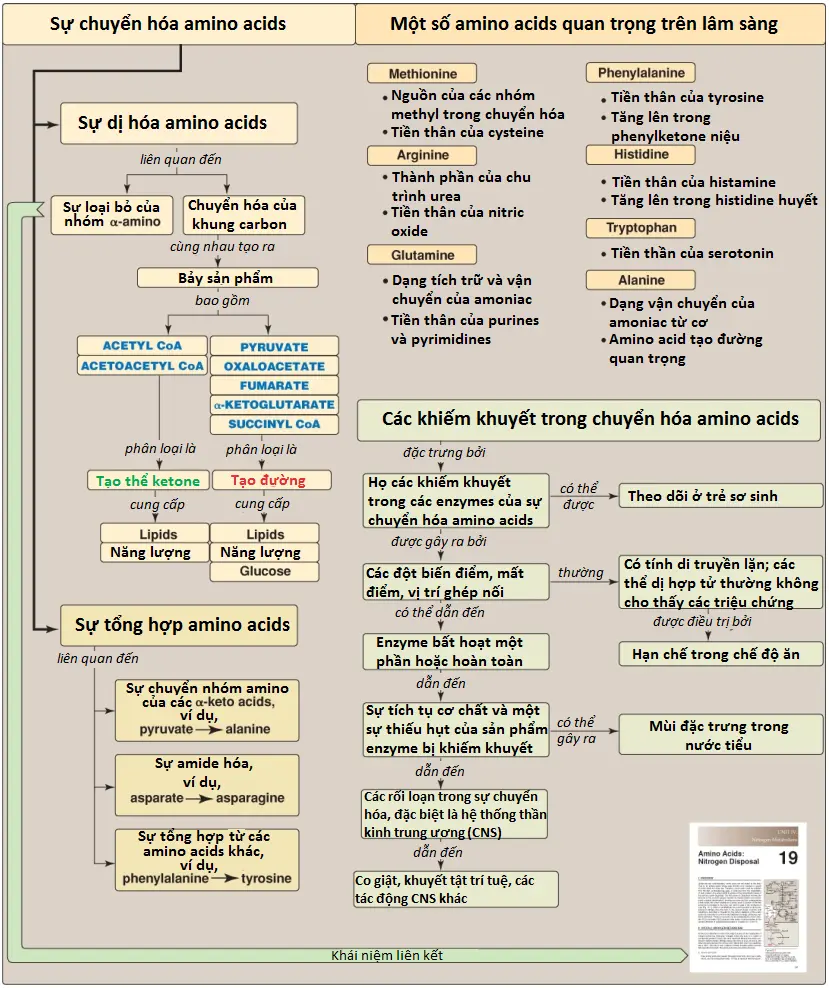
- Các amino acids mà sự chuyển hóa của chúng tạo ra pyruvate hoặc một trung gian của chu trình TCA thì chúng được gọi là các amino acids tạo đường (glucogenic) (Hình 24). Chúng có thể tạo ra sự hình thành cuối cùng của glucose trong gan và các thận. Các amino acids chỉ tạo đường là glutamine, glutamate, proline, arginine, histidine, alanine, serine, glycine, cysteine, methionine, valine, threonine, aspartate và asparagine.
- Các amino acids mà sự chuyển hóa của chúng tạo ra acetyl CoA (một cách trực tiếp mà không có pyruvate đóng vai trò như là một trung gian) hoặc acetoacetate (hoặc tiền thân của nó là acetoacetyl CoA) được gọi là các amino acids tạo thể ketone (ketogenic). Leucine và lysine là các amino acids chỉ tạo thể ketone.
- Tyrosine, phenylalanine, tryptophan và isoleucine thì vừa tạo đường, vừa tạo thể ketone.
- Các amino acids không thiết yếu (nonessential amino acids) có thể được tổng hợp từ các trung gian chuyển hóa hoặc từ các khung carbon của các amino acids thiết yếu.
- Các amino acids thiết yếu (essential amino acids) cần phải được hấp thu từ chế độ ăn. Chúng bao gồm histidine, methionine, threonine, valine, isoleucine, phenylalanine, tryptophan, leucine và lysine.
- PKU được gây ra bởi một sự thiếu hụt của PAH, enzyme giúp chuyển phenylalanine thành tyrosine. Tăng phenylalanine máu (hyperphenylalaninemia) cũng có thể được gây ra bởi các sự thiếu hụt trong các enzymes mà giúp tổng hợp hoặc tái tạo coenzyme cho PAH, là BH4. Những người mắc PKU không điều trị sẽ bị khuyết tật trí tuệ nghiêm trọng, chậm phát triển, tật đầu nhỏ, co giật và một mùi mốc đặc trưng trong nước tiểu. Điều trị liên quan đến sự kiểm soát phenylalanine trong chế độ ăn. Tyrosine trở thành một thành phần thiết yếu trong chế độ ăn đối với những người mắc PKU.
- MSUD được gây ra bởi một sự thiếu hụt một phần hoặc hoàn toàn trong BKCD, enzyme mà decarboxyl hóa các BCAAs, là leucine, isoleucine và valine. Các triệu chứng bao gồm các vấn đề về ăn uống, nôn mửa, nhiễm toan ketoacid, các thay đổi trong trương lực cơ và một mùi siro đặc trưng của nước tiểu. Nếu không điều trị, bệnh sẽ dẫn đến các vấn đề về thần kinh mà gây ra tử vong. Điều trị liên quan đến việc kiểm soát lượng BCAA ăn vào.
- Các bệnh di truyền quan trọng khác liên quan với chuyển hóa amino acid bao gồm bạch tạng, homocystine niệu, MMA, alkapton niệu, histidine huyết, tyrosine huyết và cystathionine niệu.
Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/profile.php?id=61550892771585
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/amino-acids-su-thoai-hoa-va-su-tong-hop-phan-2/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!







{kind=link}