






























VI. Sự chuyển hóa amoniac
Amoniac được sản xuất bởi tất cả các mô trong suốt quá trình chuyển hóa của nhiều hợp chất và nó được loại bỏ chủ yếu bởi sự hình thành của urea trong gan. Tuy nhiên, mức amoniac máu phải được giữ ở mức rất thấp bởi vì ngay cả khi các nồng độ tăng lên rất ít (tăng amoniac máu) thì đều gây độc với hệ thống thần kinh trung ương. Vì thế, một cơ chế được cần đến để vận chuyển nitrogen từ các mô ngoại vi đến gan cho sự loại bỏ cuối cùng dưới dạng urea đồng thời giữ các mức amoniac tuần hoàn thấp.
A. Các nguồn
Các amino acids là nguồn amoniac quan trọng nhất về mặt định lượng bởi vì hầu hết các chế độ ăn kiểu phương Tây thì giàu protein và tạo ra các amino acids dư thừa, thành phần mà đi qua gan và trải qua sự chuyển khử nhóm amino (transdeamination) (nghĩa là sự kết hợp của các phản ứng aminotransferase và GDH), tạo thành amoniac. (Chú ý: Gan dị hóa các amino acids chuỗi thẳng là chủ yếu). Tuy nhiên, các lượng amoniac đáng kể có thể thu được từ các nguồn khác.
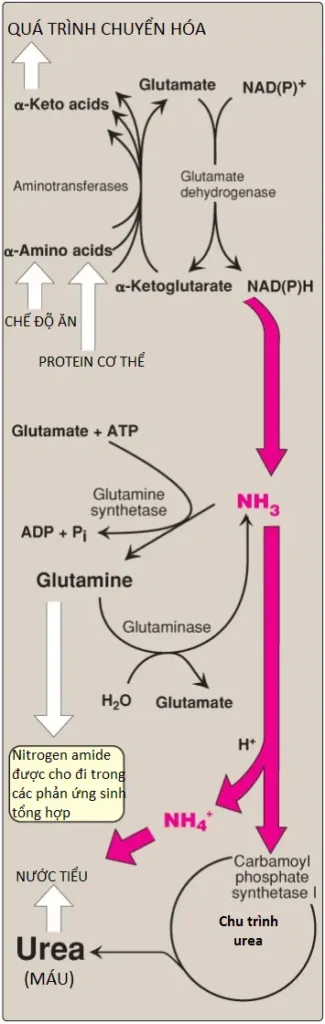
1. Glutamine: Một nguồn glutamine huyết tương quan trọng là từ sự dị hóa của BCAA (các amino acid phân nhánh [branched-chain amino acids]) trong cơ xương. Glutamine này được hấp thu bởi các tế bào của ruột, gan và các thận. Gan và các thận tạo ra amoniac từ glutamine bởi các hoạt động của glutaminase (Hình 17) và GDH. Trong các thận, hầu hết amoniac này được bài tiết vào trong nước tiểu dưới dạng NH4+, cung cấp một cơ chế quan trọng cho việc duy trì cân bằng acid-base của cơ thể thông qua sự bài tiết của các protons. Trong gan, amoniac được khử độc thành urea và được bài tiết. (Chú ý: α-ketoglutarate, sản phẩm thứ hai của GDH, là tiền thân của quá trình tạo đường trong gan và các thận). Amoniac cũng được tạo thành bởi glutaminase ruột. Các tế bào niêm mạc ruột thu được glutamine cả từ máu và từ sự tiêu hóa của protein chế độ ăn. (Chú ý: Sự chuyển hóa glutamine ruột cũng tạo thành alanine, thành phần mà được sử dụng bởi gan cho quá trình tân tạo đường và citrulline, thành phần được sử dụng bởi các thận để tổng hợp arginine).

2. Vi khuẩn đường ruột: Amoniac được hình thành từ urea bởi hoạt động của urease vi khuẩn trong lòng ruột. Amoniac này được hấp thu từ đường ruột bởi con đường tĩnh mạch cửa và hầu như được loại bỏ bởi gan thông qua sự chuyển đổi thành urea.
3. Các amines: Các amines từ chế độ ăn và các monoamines mà đóng vai trò như là các hormones hoặc các chất dẫn truyền thần kinh giúp tạo ra amoniac bởi hoạt động của monoamine oxidase.
4. Purines và pyrimidines: Trong sự dị hóa của purines và pyrimidines, các nhóm amino nối với các nguyên tử của vòng được giải phóng dưới dạng amoniac.
B. Sự vận chuyển trong hệ tuần hoàn
Mặc dù amoniac được sản xuất liên tục trong các mô nhưng nó xuất hiện ở các mức rất thấp trong máu. Điều này là do cả sự loại bỏ nhanh amoniac khỏi máu bởi gan và bởi một số mô, đặc biệt là cơ, giải phóng nitrogen của amino acid ở dạng glutamine và alanine chứ không phải là dưới dạng amoniac tự do (Hình 13, phần 2).
1. Urea: Sự hình thành của urea trong gan là chặng loại bỏ amoniac quan trọng nhất. Urea đi trong máu từ gan đến các thận, nơi mà nó đi vào trong dịch lọc của cầu thận.
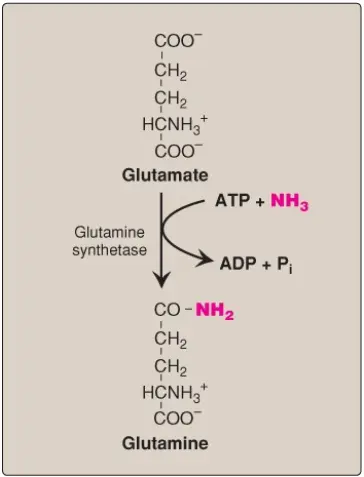
2. Glutamine: Amide này của glutamate cung cấp một sự tích trữ và dạng vận chuyển không gây độc của amoniac (Hình 18). Sự hình thành cần ATP của glutamine từ glutamate và amoniac bởi glutamine synthetase xảy ra chủ yếu trong cơ xương và gan nhưng cũng quan trọng trong hệ thống thần kinh tủng ương, nơi mà nó là cơ chế chính cho sự loại bỏ của amoniac trong não. Glutamine được tìm thấy trong huyết tương ở các nồng độ cao hơn so với các amino acids khác, một phát hiện phù hợp với chức năng vận chuyển của nó. (Chú ý: Gan giữ các mức amoniac máu thấp thông qua glutaminase, GDH và chu trình urea trong các tế bào gan quanh cửa gan [gần đường vào của máu] và thông qua glutamine synthetase như là một sự dọn dẹp amoniac trong các tế bào gan quanh tĩnh mạch trung tâm). Sự chuyển hóa amoniac được tóm tắt trong Hình 19.
C. Sự tăng ammonia máu (hyperammonemia)
Khả năng của chu trình urea của gan vượt quá tốc độ hình thành của amoniac bình thường và các mức của amoniac máu bình thường thì thấp (5 đến 35 micromol/l). Tuy nhiên, khi chức năng gan bị suy giảm, do cả các khiếm khuyết di truyền của chu trình urea hoặc bệnh gan, các mức amoniac trong máu có thể >1000 micromol/l/. Tình trạng tăng ammonia máu như vậy là một cấp cứu y khoa, bởi vì amoniac có một tác động gây độc thần kinh trực tiếp lên hệ thống thần kinh trung ương. Ví dụ, các nồng độ amoniac tăng lên trong máu gây ra các triệu chứng của nhiễm độc amoniac, là tình trạng mà bao gồm các triệu chứng run rẩy, nói líu, lơ mơ, nôn ói, phù não và nhìn mờ. Ở các nồng độ cao, ammonia có thể gây ra hôn mê và tử vong. Có hai loại tăng ammonia máu chính.
1. Mắc phải: Bệnh gan là một nguyên nhân thường gặp của tăng amoniac máu mắc phải ở người trưởng thành và có thể là do viêm gan virus hoặc do chất độc đối với gan như rượu. Xơ gan có thể gây ra sự hình thành của tuần hoàn bàng hệ quanh gan. Kết quả, máu tĩnh mạch cửa được thông nối trực tiếp vào trong tuần hoàn hệ thống và không đến gan. Vì thế, sự chuyển đổi của amoniac thành urea bị suy giảm nghiêm trọng, dẫn đến tăng các mức của amoniac.
2. Di truyền: Các sự thiếu hụt di truyền của mỗi một enzyme trong số 5 enzymes của chu trình urea (và của NAGS) đã được mô tả, với một tỷ lệ chung là khoảng 1:25,000 đứa trẻ sinh ra. Sự thiếu hụt OTC được liên kết với NST X, chủ yếu ảnh hưởng đến nam giới, mặc dù nữ giới mang gene bệnh có thể có triệu chứng. Tất cả các rối loạn chu trình urea khác tuân theo một kiểu di truyền lặn trên NST thường. Trong mỗi trường hợp, sự suy yếu quá trình tổng hợp urea dẫn đến tăng amoniac máu trong suốt các tuần đầu tiên sau sinh. Các sự kết hợp của các triệu chứng khác nhau thường gặp trong tăng amoniac máu (run rẩy, nói líu, lơ mơ, nôn ói, phù não, nhìn mờ, các khiếm khuyết phát triển và trí tuệ) cũng có thể được quan sát thấy trong các tình trạng suy giảm của chu trình urea. Chẩn đoán dựa trên các triệu chứng, các xét nghiệm và các test di truyền. Trong lịch sử, các khiếm khuyết chu trình urea bẩm sinh có một tỷ lệ bệnh tật (các biểu hiện thần kinh) và tỷ lệ tử vong cao. Thông tin thêm về các sự thiếu hụt của chu trình urea cụ thể được tổng hợp trong các phần sau.
a. Sự thiếu hụt ornithine transcarbamylase: Sự thiếu hụt OTC là rối loạn chu trình urea thường gặp nhất. Các kết quả xét nghiệm đặc hiệu bao gồm một sự giảm trong phản ứng và trong các sản phẩm bên dưới vị trí enzyme thiếu hụt ,là citrulline và arginine. Đáng chú ý, một sự tăng lên trong các mức orotic acid cũng có thể được phát hiện. Carbamoyl phosphate, một trong số các cơ chất của OTC, thay vào đó sẽ trở thành một cơ chất cho sự sinh tổng hợp pyrimidine, đi vào trong con đường bên dưới của phản ứng điều hòa (xem các bài viết tiếp theo nhé). Kết quả, orotic acid là một trung gian được sản xuất quá mức của con đường sinh tổng hợp pyrimidine. (Chú ý: Orotic acid tăng lên cũng được quan sát thấy trong nhiễm toan nước tiểu do orotic acid di truyền (hereditary orotic aciduria), tình trạng này là do sự thiếu hụt enzyme sinh tổng hợp pyrimidine trong UMP synthase [UMPS]. Cùng với xét nghiệm di truyền, sự thiếu hụt OTC có thể được chẩn đoán phân biệt với sự thiếu hụt UMPS dựa trên các triệu chứng khác. Tăng amoniac máu là một triệu chứng của sự thiếu hụt OTC nhưng không phải là triệu chứng của sự thiếu hụt UMPS; thay vào đó, thiếu máu hồng cầu khổng lồ có thể là một triệu chứng của sự thiếu hụt UMPS).
b. Sự thiếu hụt argininosuccinate synthetase: Sự thiếu hụt này còn được gọi là tăng citrulline máu type 1 (citrullinemia type 1), bởi vì có một sự tích tụ của cơ chất cho phản ứng, là citrulline, trong máu và nước tiểu. Có thể có một dạng cấp sơ sinh (cổ điển), một dạng khởi phát trễ nhẹ hơn, một dạng mà bắt đầu trong suốt hoặc sau thai kỳ, và một dạng không có triệu chứng. Trong thể cấp sơ sinh, citrulline có thể được phát hiện như là một phần của kết quả theo dõi sơ sinh. Sự phát hiện này là cần thiết để ngăn chặn tình trạng tăng amoniac máu và tổn thương não.
c. Sự thiếu hụt argininosuccinate lyase: Trong tình trạng thiếu hụt argininosuccinate lyase, có một sự tích tụ của cơ chất cho phản ứng, là argininosuccinate, trong nước tiểu, gây ra tình trạng nhiễm toan nước tiểu do argininosuccinic (argininosuccinic aciduria). Điều này có ý nghĩa chẩn đoán và là một phần của theo dõi sơ sinh. Trong các dạng nghiêm trọng và khởi phát trễ hơn của sự thiếu hụt, nhiễm toan nước tiểu có thể liên quan với các bất thường thần kinh, các sự chậm phát triển và sự suy giảm nhận thức.
d. Tình trạng thiếu hụt arginase-I: Trong tình trạng thiếu hụt arginase-I, có một sự tích tụ của cơ chất cho phản ứng, là arginine, trong máu và trong nước tiểu, và thường được gọi là tăng arginine máu (hyperargininemia hay argininemia). Tăng amoniac máu được quan sát thấy với sự thiếu hụt arginase thường ít nghiêm trọng hơn bởi vì arginine chứa 2 nitrogens thải (waste nitrogens) và có thể được bài tiết trong nước tiểu. Do đó, các bệnh nhân mắc tình trạng thiếu hụt này có thể khỏe mạnh khi sinh và có sự phát triển bình thường trong suốt 1 đến 3 năm đầu. Sau thời gian này, các triệu chứng đầu tiên của tình trạng thiếu hụt arginase có thể xuất hiện với sự chậm trễ rõ ràng trong quá trình phát triển, mất các dấu mốc phát triển và khiếm khuyết trí tuệ. Tăng amoniac máu có thể là theo từng đợt, liên quan với các bữa ăn giàu protein hoặc các giai đoạn stress, như bênh tật hoặc đói.
e. Sự thiếu hụt của N-acetylglutamate synthase: Giống như sự thiếu hụt aginase-I, một sự thiếu hụt trong NAGS có thể gây ra các sự chậm trễ trong quá trình phát triển và sự khiếm khuyết trị tuệ. Các dạng ít nghiêm trọng hơn có thể xuất hiện từng đợt trong đời sống, liên quan với các bữa ăn giàu protein, stress hoặc đói. Carglumic acid là một liệu pháp đã được chứng minh bởi FDA đối với tình trạng thiếu hụt NAGS. Nó là một dạng tổng hợp của NAG, là chất hoạt hóa dị lập thể dương của carbamoyl phosphate synthetase I.
f. Sự điều trị đối với tăng ammonia máu: Sự điều trị đối với các sự thiếu hụt của enzyme chu trình urea liên quan đến cả việc hạn chế protein ăn vào trong chế độ ăn với đủ lượng calories để ngăn cản sự dị hóa protein và việc loại bỏ amoniac dư thừa trong máu. Điều này có thể thay đổi phụ thuộc vào tình trạng thiếu hụt enzyme và độ nghiêm trọng của khiếm khuyết. Các bệnh nhân sẽ gắn liền với chế độ ăn ít protein, với các mức protein tối thiểu cần để duy trì sức khỏe tốt. Điều này có thể thay đổi phụ thuộc vào tuổi và trọng lượng của bệnh nhân. Các đồ uống với các công thức đặc biệt và/hoặc các thực phẩm y khoa có thể được mua để sử dụng, mà trong đó các mức protein đã được điều chỉnh cho các nhu cầu của bệnh nhân. Các thuốc “dọn dẹp” nitrogen (nitrogen-scavenging medications), bao gồm các acids thơm benzoate và phenylbutyrate, có thể làm giảm các mức amoniac trong máu. Benzoate kết hợp với glycine để hình thành nên hippurate. Phenylbutyrate được chuyển thành phenylacetate và kết hợp với glutamine để hình thành nên phenylacetylglutamine (Hình 20). Cả hai sản phẩm cuối cùng, hippurate và phenylacetylglutamine, thì đều dễ dàng được bài tiết trong nước tiểu. Sự bài tiết kết hợp của glycine và glutamine và sự sinh tổng hợp kế tiếp của chúng làm giảm một cách hiệu quả các mức amoniac và nguy cơ tăng amoniac máu. Trong suốt tình trạng tăng amoniac máu nặng, các bệnh nhân cũng có thể cần thẩm phân máu, truyền dịch tĩnh mạch hoặc các sự điều trị khác để nhanh chóng làm giảm các mức amoniac máu và ngăn ngừa tổn thương não vĩnh viễn.

VII. Tóm tắt loạt bài viết

- Nitrogen đi vào trong cơ thể trong nhiều loại hợp chất khác nhau có trong thực phẩm, quan trọng nhất là các amino acids chứa trong protein chế độ ăn.
- Nitrogen rời cơ thể dưới dạng urea, amoniac và các sản phẩm khác có nguồn gốc từ sự chuyển hóa amino acid (Hình 21).
- Các amino acids tự do trong cơ thể được tạo ra bằng sự thủy phân của protein chế độ ăn bởi các proteases được hoạt hóa từ dạng zymogen (tiền enzyme) của chúng trong dạ dày và ruột, sự thoái hóa của các proteins mô và sự tổng hợp mới. Hồ amino acid (amino acid pool) này được tiêu thụ trong sự tổng hợp protein của cơ thể, được chuyển hóa để sinh năng lượng hoặc các thành viên của nó được sử dụng như là các tiền thân cho các hợp chất chứa nitrogen khác.
- Các amino acids tự do từ sự tiêu hóa được hấp thu bởi các tế bào niêm mạc ruột thông qua sự vận chuyển tích cực thứ phát phụ thuộc natri. Các peptides nhỏ được hấp thu thông qua sự vận chuyển liên kết với proton.
- Protein của cơ thể được thoái hóa và tái tổng hợp một cách đồng thời, một quá trình được gọi là sự thay thế protein (protein turnover). Nồng độ của một protein tế bào có thể được xác định bởi sự điều hòa tổng hợp và thoái hóa của nó. Ub-proteasome chọn lọc, trong bào tương, phụ thuộc ATP và các acid hydrolases của lysosome không phụ thuộc ATP và tương đối không chọn lọc là hai hệ thống enzyme chính mà chịu trách nhiệm cho sự thoái hóa các proteins.
- Nitrogen không thể được tích trữ và các amino acids dư thừa của các nhu cầu sinh tổng hợp của tế bào nhanh chóng được thoái hóa. Giai đoạn dị hóa đầu tiên liên quan đến sự chuyển các nhóm α-amino thông qua sự chuyển nhóm amino (transamination) bởi aminotransferases (transaminases) phụ thuộc pyridoxal phosphate, được theo sau bởi sự khử amino oxy hóa của glutamate bởi GDH, hình thành nên amoniac và các α-keto acids tương ứng.
- Một phần của amoniac tự do được bài tiết trong nước tiểu. Một ít amoniac được sử dụng trong việc chuyển đổi glutamate thành glutamine cho sự vận chuyển an toàn, nhưng hầu hết được sử dụng trong sự tổng hợp urea của gan, là chặng quan trọng nhất về mặt định lượng để loại bỏ nitrogen khỏi cơ thể. Alanine cũng mang nitrogen đến gan để loại bỏ dưới dạng urea.
- Hai nguyên nhân chính của tình trạng tăng amoniac máu (với các tác động thần kinh của nó) là bệnh gan mắc phải và các sự thiếu hụt bẩm sinh của các enzymes của chu trình urea như tình trạng thiếu hụt OTC liên kết NST X.
Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/profile.php?id=61550892771585
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/amino-acids-su-loai-bo-nitrogen-nito-phan-3/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!







{kind=link}