





























I. Tổng quan
Ngoài đóng vai trò như là các thành phần cấu trúc cho các proteins, các amino acids cũng là tiền thân của nhiều hợp chất chứa nitrogen (N) mà có các chức năng sinh lý quan trọng (Hình 1). Các phân tử này bao gồm porphyrins, các chất dẫn truyền thần kinh, các hormones, purines và pyrimidines. (Chú ý: Xem các bài viết trước cho sự tổng hợp của nitric oxide từ arginine).
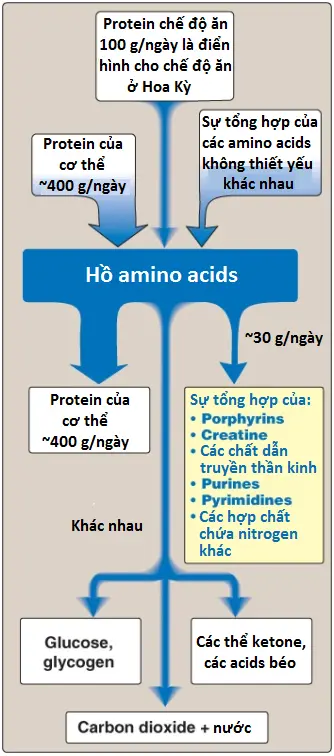
II. Chuyển hóa porphyrins
Porphyrins là các phân tử vòng mà dễ dàng liên kết với các ion sắt, thường là sắt II (Fe2+) hoặc sắt III (Fe3+). Porphyrin-kim loại phổ biến nhất ở người là heme, là thành phần chứa một Fe2+ liên kết ở trung tâm của vòng tetrapyrrole của protoporphyrin IX. Heme là nhóm phụ đối với hemoglobin (Hb), myoglobin, cytochromes, bao gồm cả hệ thống cytochrome P450 (CYP) monooxygenase, catalase, nitric oxide synthase và peroxidase. Các proteins heme này nhanh chóng được tổng hợp và thoái hóa. Ví dụ, 6 đến 7 grams Hb được tổng hợp mỗi ngày để thay thế cho sự mất heme thông qua sự thay thế bình thường của hồng cầu. Sự tổng hợp và sự thoái hóa của porphyrins liên quan và sự tái chế của sắt được phối hợp với sự thay thế của các proteins heme.
A. Cấu trúc
Porphyrins là các phân tử phẳng vòng mà được hình thành bởi sự liên kết của 4 vòng pyrrole thông qua các cầu nối methenyl (Hình 2). Ba đặc điểm cấu trúc của các phân tử này thì cần thiết để có thể hiểu được tầm quan trọng y khoa của chúng:

1. Các chuỗi bên: Các porphyrins khác nhau thì sẽ khác nhau về bản chất của các chuỗi bên mà bám vào mỗi trong số 4 vòng pyrrole. Uroporphyrin chứa các chuỗi bên acetate (-CH2-COO-) và propionate (-CH2-CH2-COO-); coproporphyrin chứa các nhóm methyl (-CH3) và propionate; và protoporphyrin IX (và heme b là heme phổ biến nhất) chứa các nhóm vinyl (-CH=CH2), methyl và propionate. (Chú ý: Các nhóm methyl và vinyl được tạo ra một cách tương ứng bởi sự decarboxyl hóa các chuỗi bên acetate và propionate).
2. Sự phân bố chuỗi bên: Các chuỗi bên của porphyrins thì có thể được sắp xếp quanh nhân tetrapyrrole theo 4 cách khác nhau, được ký hiệu bởi các chữ số La Mã từ I đến IV. Chỉ porphyrins type III, mà chứa một sự thay thế bất đối xứng trên vòng D (Hình 2), là quan trọng về mặt sinh lý ở người. (Chú ý: Protoporphyrin IX là một thành viên của type III).
3. Porphyrinogens: Các tiền thân porphyrin này (như uroporphyrinogen) tồn tại ở một dạng khử hóa học, không màu và đóng vai trò như là các trung gian giữa porphobilinogen (PBG) và các protoporphyrins oxy hóa có màu trong sinh tổng hợp heme.
B. Sinh tổng hợp heme
Các vị trí chính của sự tổng hợp heme là gan và các tế bào sinh hồng cầu của tủy xương. Trong gan, nơi tổng hợp một số proteins heme (cụ thể là các proteins CYP), mức độ tổng hợp heme thì có sự thay đổi lớn, đáp ứng với các sự thay đổi trong dự trữ heme của tế bào được gây ra bởi các nhu cầu biến động đối với các proteins heme. Ngược lại, sự tổng hợp heme trong các tế bào sinh hồng cầu, mà hoạt động trong sự tổng hợp Hb, thì tương đối hằng định và phù hợp với mức độ tổng hợp globin. (Chú ý: Hơn 85% tất cả sự tổng hợp heme là xảy ra trong mô sinh hồng cầu. Các tế bào hồng cầu trưởng thành (RBCs) thiếu ty thể và không thể tổng hợp heme). Phản ứng khởi động và 3 bước cuối cùng trong sự hình thành của porphyrins là xảy ra trong ty thể, ngược lại, các bước trung gian của con đường sinh tổng hợp là xảy ra trong bào tương. (Chú ý: Hình 8 [phần 2] sẽ tóm tắt quá trình tổng hợp heme).
1. Sự hình thành δ-Aminolevulinic acid: Tất cả nguyên tử carbon và nitrogen của porphyrin đều được cung cấp bởi glycine (một amino acid không thiết yếu) và succinyl coenzyme A (một trung gian của chu trình tricarboxylic acid) mà ngưng tụ để hình thành nên δ-Aminolevulinic acid (ALA) trong một phản ứng được xúc tác bởi ALA synthase ([ALAS], Hình 3). Phản ứng này cần pyridoxal phosphate ([PLP]) như là một coenzyme và là bước cam kết và giới hạn tốc độ trong sinh tổng hợp porphyrin. (Chú ý: Có hai isoforms (dạng) ALAS, mỗi trong số đó được sản xuất bởi các gen khác nhau và được kiểm soát bởi các cơ chế khác nhau. ALAS1 được tìm thấy trong tất cả các mô, ngược lại ALAS2 thì đặc hiệu cho mô sinh hồng cầu. Các đột biến mất chức năng trong ALAS2 gây ra thiếu máu nguyên hồng cầu liên kết NST X và tình trạng quá tải sắt).
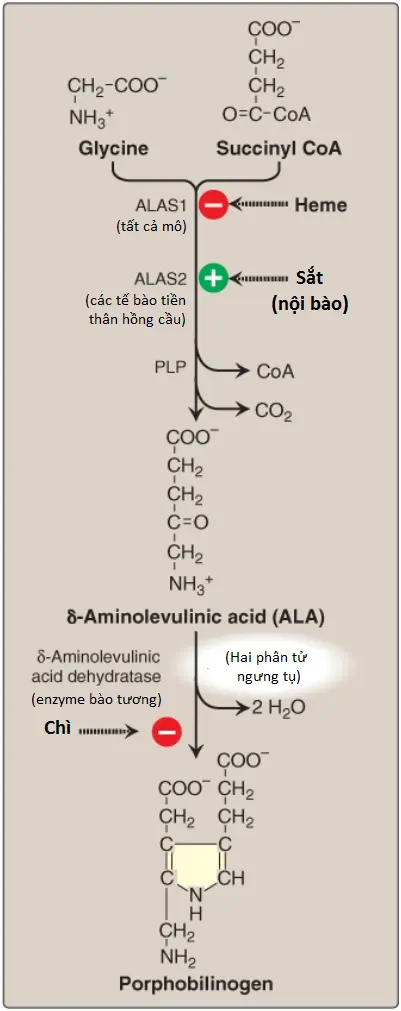
a. Các tác động của heme (hemin): Khi sự sản xuất porphyrin vượt quá sự có mặt sẵn của apoproteins mà cần đến nó, heme sẽ tích tụ và được chuyển thành hemin bởi sự oxy hóa của Fe2+ thành Fe3+. Hemin làm giảm lượng (và vì thế, hoạt động) của ALAS1 bằng cách ngăn chặn sự dịch mã gen của nó, làm tăng sự thoái hóa RNA thông tin của nó và làm giảm sự nhập của enzyme vào trong ty thể. (Chú ý: Trong các tế bào sinh hồng cầu, ALAS2 được kiểm soát bởi sự có mặt sẵn của sắt nội bào).
b. Các tác động của thuốc: Sự sử dụng bất cứ thuốc nào trong số nhiều thuốc (và các hóa chất lạ môi trường, xuất hiện trong một số thức ăn, mỹ phẩm và các sản phẩm thương mại) gây ra sự tăng lên đáng kể trong hoạt động ALAS1 của gan. Các phân tử này được chuyển hóa bởi hệ thống CYP monooxygenase tiểu thể (microsomal CYP monooxygenase system), một hệ thống heme protein oxidase (heme protein oxidase system) được tìm thấy trong gan. Trong đáp ứng với các thuốc này, sự tổng hợp của CYP proteins tăng lên, dẫn đến một sự tăng cường tiêu thụ heme, một thành phần của các proteins này. Điều này, cuối cùng, sẽ gây ra một sự giảm trong nồng độ của heme tự do hay heme không liên kết trong các tế bào gan. Nồng độ heme không liên kết nội bào thấp hơn dẫn đến một sự tăng lên trong tổng hợp ALAS1 và tạo ra một sự tăng lên tương ứng trong sự tổng hợp của ALA.
2. Sự hình thành của porphobilinogen: Sự ngưng tụ trong bào tương của hai ALA để hình thành nên PBG bởi ALA dehydratase chứa kẽm (PBG synthase) thì cực kỳ nhạy cảm với sự ức chế bởi các ions kim loại nặng (như chì), thành phần mà dẫn đến thay thế kẽm (Hình 3). Sự ức chế này phần nào chịu trách nhiệm cho sự tăng lên của ALA và thiếu máu được gây ra bởi ngộ độc chì.
3. Sự hình thành uroporphyrinogen: Sự ngưng tụ của 4 phân tử PBG, được xúc tác bởi hydroxymethylbilane synthase, tạo ra tetrapyrrole hydroxymethylbilane dạng thẳng. Một sự thiếu hụt trong enzyme này gây ra một rối loạn chuyển hóa porphyrin từng đợt cấp tính (acute intermittent porphyria) (AIP, Hình 4, cũng xem phần 2 để có nhiều chi tiết hơn về các dạng khác nhau của các rối loạn chuyển hóa porphyrin [porphyria]). Uroporphyrinogen III synthase đóng vòng và isomer hóa (đồng phân hóa) hydroxymethylbilane để tạo thành uroporphyrinogen III bất đối xứng. Một sự thiếu hụt trong enzyme này gây ra rối loạn chuyển hóa porphyrin hồng cầu bẩm sinh (congenital erythropoietic porphyria – CEP). Uroporphyrinogen III trải qua sự decarboxyl hóa của các nhóm acetate của nó bởi uroporphyrinogen III decarboxylase (UROD), tạo thành coproporphyrinogen III. Một sự thiếu hụt trong enzyme này gây ra rối loạn chuyển hóa porphyrin biểu hiện da muộn (porphyria cutanea tarda – PCT). Ba phản ứng này xảy ra trong bào tương.

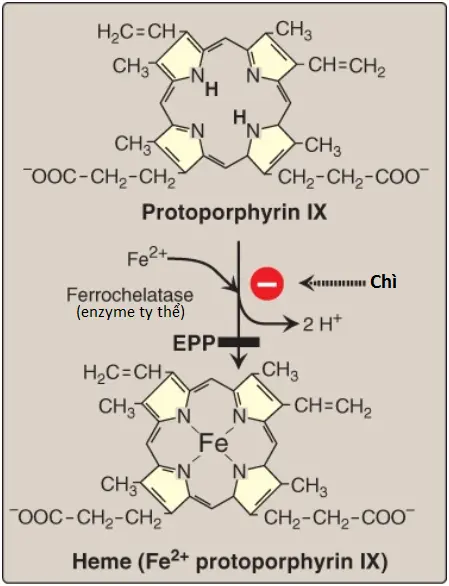
4. Sự hình thành heme: Coproporphyrinogen III đi vào trong ty thể, và hai chuỗi bên propionate được decarboxyl hóa bởi coproporphyrinogen III oxidase thành các nhóm vinyl, tạo thành protoporphyrinogen IX. Một sự thiếu hụt trong enzyme này gây ra coproporphyria di truyền (hereditary coproporphyria – HCP). Protoporphyrinogen IX được oxy hóa bởi protoporphyrinogen oxidase thành protoporphyrin IX. Một sự thiếu hụt trong enzyme này gây ra porphyria kết hợp (variegate porphyria – VP). Sự xuất hiện của sắt (dưới dạng Fe2+) trong protoporphyrin IX sẽ tạo ra heme. Bước này cũng có thể tự phát, nhưng tốc độ được tăng cường bởi ferrochelatase, một enzyme mà, giống như ALA dehydratase, bị ức chế bởi chì (Hình 5). Một sự thiếu hụt trong enzyme này gây ra protoporphyria hồng cầu (erythropoietic protoporphyria – EPP).
Áp dụng lâm sàng 21.1: Ngộ độc chì
Ngộ độc chì là một sự tích tụ chì trong cơ thể trong một khoảng thời gian nhiều tháng đến nhiều năm. Các nguồn chì thường gặp bao gồm sơn có chứa chì và bụi sơn hoặc vảy sơn trong các tòa nhà cũ; chì trong các ống nước gia dụng cũng có thể nhiễm vào trong nước uống. Phơi nhiễm cũng có thể xảy ra thông qua hít phải, tiếp xúc với da hay các màng niêm mạc hoặc ăn phải. Chì có một vị ngọt và phơi nhiễm ăn phải là một mối quan tâm đặc biệt đối với trẻ mới sinh và trẻ nhỏ. Các triệu chứng của ngộ độc chì có thể bao gồm chậm phát triển, khiếm khuyết khả năng học tập và chỉ số IQ thấp, đau bụng, táo bón, các thay đổi thần kinh và khó chịu. Các mức chì rất cao có thể gây tử vong. Chì ức chế ALA dehydratase và ferrochelatase, cả hai enzyme đều liên quan đến sự tổng hợp heme và vì thế gây ra một sự giảm trong sự tổng hợp heme. Hơn thế nữa, các mức cao chì làm giảm khả năng sử dụng sắt. Điều này gây ra sự tăng sử dụng kẽm (thay vì sắt) như là cơ chất cho sự tạo chelate đối với protoporphyrin IX bởi ferrochelatase. Kết quả, các bệnh nhân mắc ngộ độc chì có thể có biểu hiện thiếu máu và tăng các mức protoporphyrin kẽm. Sự tăng lên trong ALA có thể là gây độc đối với các tế bào thần kinh. Chì cũng có thể đi qua hàng rào máu-não và gây độc thần kinh. Điều trị thông thường là loại bỏ nguồn phơi nhiễm đối với chì nhưng trong các trường hợp ngộ độc chì nặng (nhiều hơn 45 μg/dl được đo trong huyết thanh), các chất tạo chelate với kim loại hóa trị hai như succimer (DMSA, 2,3-dimercaptosuccinic acid), calcium disodium ethylenediaminetetraacetic acid (EDTA) hoặc các chất khác cũng có thể được sử dụng để loại bỏ các ions chì dư thừa khỏi máu.
Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/profile.php?id=61550892771585
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/amino-acids-su-thoai-hoa-va-su-tong-hop-phan-3/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!







{kind=link}