





























I. Tổng quan
Sự thoái hóa amino acid liên quan đến sự loại bỏ của nhóm α-amino, được theo sau bởi sự dị hóa của các α-keto acids tạo thanh (các khung carbon). Các con đường thoái hóa của các amino acids khác nhau hội tụ lại để hình thành nên 7 sản phẩm trung gian: oxaloacetate, pyruvate, α-ketoglutarate, fumarate, succinyl coenzyme A (CoA), acetyl CoA và acetoacetate. Các sản phẩm đi vào trong các con đường chuyển hóa trung gian một cách trực tiếp, tạo ra sự tổng hợp glucose, các thể ketone hay lipids hoặc tạo ra sự sản xuất năng lượng thông qua sự oxy hóa chúng thành carbon dioxide (CO2) bởi chu trình tricarboxylic acid (TCA cycle). Hình 1 cung cấp một cái nhìn tổng quan về các con đường này, với một sự tổng hợp chi tiết hơn trong phần sau của loạt bài viết. Các amino acids không thiết yếu (Hình 2) có thể được tổng hợp với các lượng đầy đủ từ các trung gian của quá trình chuyển hóa, hoặc như trong trường hợp của cysteine và tyrosine, từ các amino acids thiết yếu. Ngược lại, bởi vì các amino acids thiết yếu không thể được tổng hợp (hoặc tổng hợp với các lượng đầy đủ) bởi con người nên chúng phải được hấp thu từ chế độ ăn để cho sự tổng hợp protein bình thường được diễn ra. Các khiếm khuyết di truyền trong các con đường chuyển hóa amino acid có thể gây ra tình trạng bệnh nghiêm trọng.
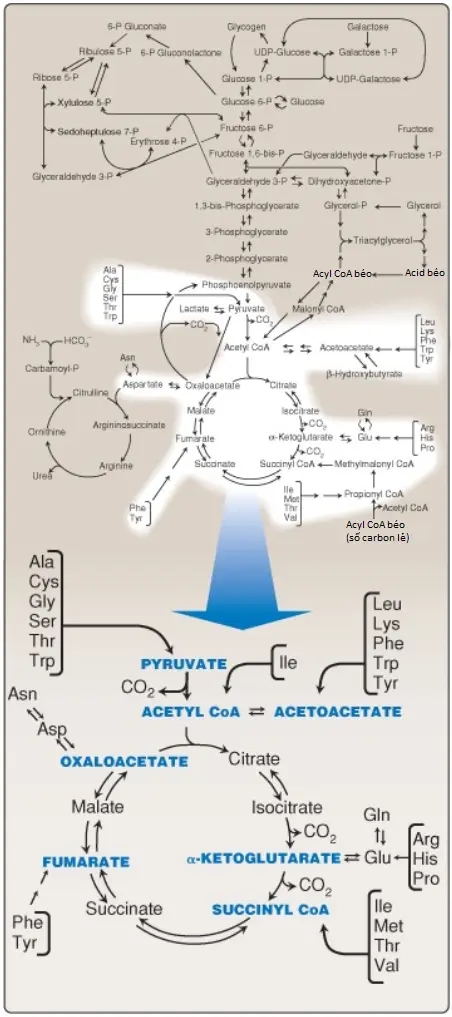
II. Các amino acids tạo đường và tạo thể ketone
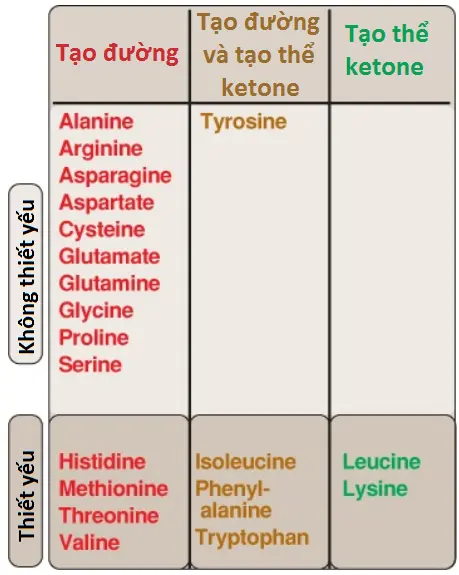
Các amino acids có thể được phân loại thành tạo đường, tạo thể ketone hoặc cả hai, dựa trên việc chất nào trong 7 trung gian được sản xuất trong suốt quá trình dị hóa của chúng (xem Hình 2).

A. Các amino acids tạo đường
Các amino acids mà quá trình chuyển hóa của chúng tạo ra pyruvate hoặc một trong số các trung gian của chu trình TCA thì được gọi là các amino acids tạo đường (glucogenic). Bởi vì các trung gian này là các cơ chất cho quá trình tân tạo đường (gluconeogenesis) nên chúng có thể gây ra sự tổng hợp cuối cùng của glucose trong gan và thận.
B. Các amino acids tạo thể ketone
Các amino acids mà sự chuyển hóa của chúng tạo ra acetyl CoA (một cách trực tiếp, mà không có pyruvate đóng vai trò như là một trung gian) hoặc acetoacetate (hoặc tiền thân của nó là acetoacetyl CoA) được gọi là các amino acids tạo thể ketone (ketogenic) (xem Hình 2). Acetoacetate là một trong các thể ketone, các thể ketone còn lại gồm 3-hydroxybuyrate và acetone. Chỉ leucine và lysine là các amino acids hoàn toàn tạo thể ketone mà được tìm thấy trong các protein. Các khung carbon của chúng không phải là các cơ chất cho quá trình tân tạo đường và vì thế, không thể tạo ra sự tổng hợp glucose cuối cùng.
III. Sự dị hóa khung carbon của amino acid
Các con đường mà bằng cách đó các amino acids được dị hóa thì được tổ chức một cách phù hợp mà theo đó một (hoặc nhiều hơn) trong số 7 trung gian được liệt kê ở trên được tạo ra từ một amino acid nhất định.
A. Các amino acids mà hình thành nên oxaloacetate
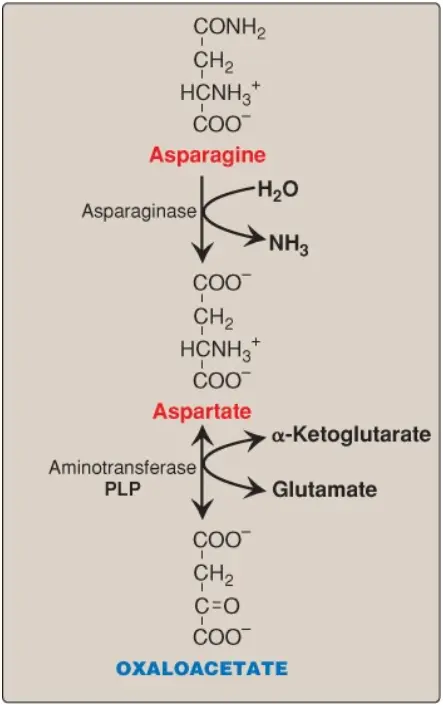
Asparagine được thủy phân bởi asparaginase, giải phóng ra amoniac và aspartate (Hình 3). Aspartate được chuyển thành ketoacid tương ứng của chúng bởi sự chuyển nhóm amino để hình thành nên oxaloacetate (xem Hình 3). (Chú ý: Một số tế bào của bệnh bạch cầu thì không thể tổng hợp đủ asparagine để hỗ trợ cho sự tăng trưởng của chúng. Điều này khiến asparagine trở thành một amino acid thiết yếu đối với các tế bào này, do đó, chúng cần asparagine trong máu. Asparaginase, là enzyme thủy phân asparagine thành aspartate, có thể được sử dụng để điều trị bệnh bạch cầu. Asparaginase làm giảm mức asparagine trong huyết tương, bằng cách đó, tước đoạt đi chất dinh dưỡng cần thiết cho các tế bào ung thư).
B. Các amino acids mà giúp hình thành nên α-ketoglutarate thông qua glutamate
1. Glutamine: Amino acid này được thủy phân thành glutamate và amoniac bởi enzyme glutaminase. Glutamate được chuyển thành α-ketoglutarate bởi sự chuyển nhóm amino hoặc thông qua sự khử amino oxy hóa bởi glutamate dehydrogenase.
2. Proline: Amino acid này được oxy hóa thành glutamate. Glutamate được chuyển nhóm amino hoặc khử amino oxy hóa để hình thành nên α-ketoglutarate.
3. Arginine: Amino acid này được thủy phân bởi arginase để tạo ra ornithine (và urea). (Chú ý: Phản ứng xảy ra chủ yếu trong gan như là một phần của chu trình urea). Ornithine được chuyển tiếp theo thành α-ketoglutarate, với glutamate semialdehyde như là một trung gian.
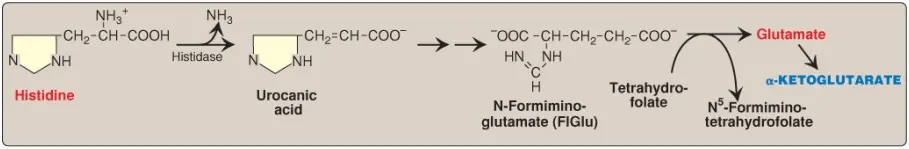
4. Histidine: Histidine được khử amino oxy hóa bởi histidase thành urocanic acid, là chất hình thành nên N-formiminoglutamate tiếp theo ([FIGLU], Hình 4). FIGLU cho đi nhóm formimino đến tetrahydrofolate (TFH), còn lại glutamate, là chất được thoái hóa như được mô tả ở trên. Một sự thiếu hụt histidase sẽ tạo ra khiếm khuyết bẩm sinh tương đối nhẹ của quá trình chuyển hóa là tăng histidine máu (histidinemia), được đặc trưng bởi sự tăng các mức histidine trong máu và trong nước tiểu. (Chú ý: Những người bị thiếu hụt folic acid sẽ làm tăng lượng FIGLU được bài tiết trong nước tiểu, đặc biệt là sau khi tiêu hóa lượng lớn histidine. Xét nghiệm bài tiết FIGLU được sử dụng để chẩn đoán một sự thiếu hụt trong folic acid. Xem các bài viết tiếp theo để hiểu về sự chuyển hóa folic acid, THF và sự chuyển hóa một carbon [one-carbon metabolism]).
C. Các amino acids hình thành nên pyruvate
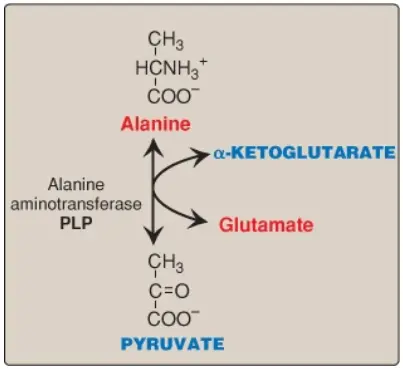
1. Alanine: Amino acid này làm mất nhóm amino của nó bởi sự chuyển nhóm amino để hình thành nên pyruvate (Hình 5). (Chú ý: Sự dị hóa tryptophan tạo ra alanine và vì thế, tạo ra pyruvate [xem thêm các phần sau]).
2. Serine: Amino acid này có thể được chuyển thành glycine khi THF trở thành N5, N10-methylenetetrahydrofolate (N5,N10-MTHF), như được thể hiện trong Hình 6A. Serine cũng có thể được chuyển thành pyruvate (xem Hình 6B).
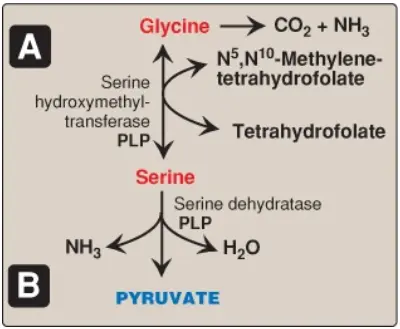
3. Glycine: Amino acid này có thể được chuyển thành serine bởi sự bổ sung có thể đảo ngược được của một nhóm methylene từ N5,N10-MTHF (xem Hình 6A) hoặc được oxy hóa thành CO2 và amoniac bởi hệ thống phân tách glycine. Glycine có thể được khử amino thành glyoxylate (bởi một D-amino acid oxidase), chất mà có thể được oxy hóa thành oxalate hoặc được chuyển nhóm amino thành glycine. Sự thiếu hụt của transaminase trong các peroxisomes gan gây ra sự sản xuất quá mức oxalate, sự hình thành của các sỏi oxalate và tình trạng tổn thương thận (oxalate niệu nguyên phát type 1).
4. Cysteine: Amino acid chứa lưu huỳnh này trải qua sự khử lưu huỳnh để thu được pyruvate. (Chú ý: Sulfate được giải phóng có thể được sử dụng để tổng hợp 3’-phosphoadenosine-5’-phosphosulfate [PAPS], một chất cho sulfate được hoạt hóa cho nhiều chất nhận khác nhau). Cysteine cũng có thể được oxy hóa thành dẫn xuất disulfide của nó, là cystine.
5. Threonine: Amino acid được chuyển thành pyruvate trong hầu hết các sinh vật nhưng là một con đường chuyển hóa thứ yếu (tối ưu nhất) ở con người.
D. Các amino acids hình thành fumarate
1. Phenylalanine và tyrosine: Sự hydroxyl hóa của phenylalanine tạo thành tyrosine (Hình 7). Phản ứng không thể đảo ngược này, được xúc tác bởi phenylalanine hydrolase (PAH) cần tetrahydrobiopterin (BH4), khởi động quá trình dị hóa của phenylalanine. Vì thế, chuyển hóa phenylalanine và chuyển hóa tyrosine được kết hợp lại, dẫn đến sự hình thành cuối cùng của fumarate và acetoacetate. Do đó, phenylalanine và tyrosine là các amino acids vừa tạo đường vừa tạo thể ketone.
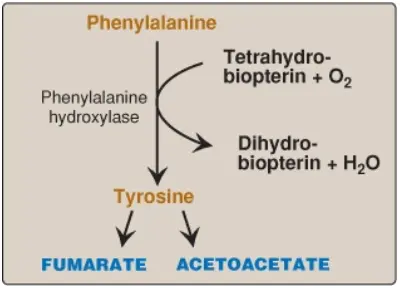
2. Các sự thiếu hụt di truyền: Các sự thiếu hụt di truyền trong các enzymes của sự chuyển hóa phenylalanine và tyrosine dẫn đến các bệnh phenylketone niệu (phenylketonuria – PKU), tăng tyrosine máu (tyrosinemia) và alkapton niệu (alkaptonuria) cũng như là bệnh bạch tạng.
E. Các amino acids mà giúp hình thành nên succinyl CoA: methionine
Methionine là một trong số bốn amino acids mà giúp hình thành nên succinyl CoA. Amino acid chứa lưu huỳnh này xứng đáng được chú ý đặc biệt bởi vì nó được chuyển thành S-adenosylmethionine (SAM), chất cho nhóm methyl chính trong quá trình chuyển hóa một carbon (Hình 8). Methionine cũng là nguồn tạo ra homocysteine (Hcy), một sản phẩm chuyển hóa liên quan với bệnh xơ vữa động mạch và huyết khối (thrombosis).
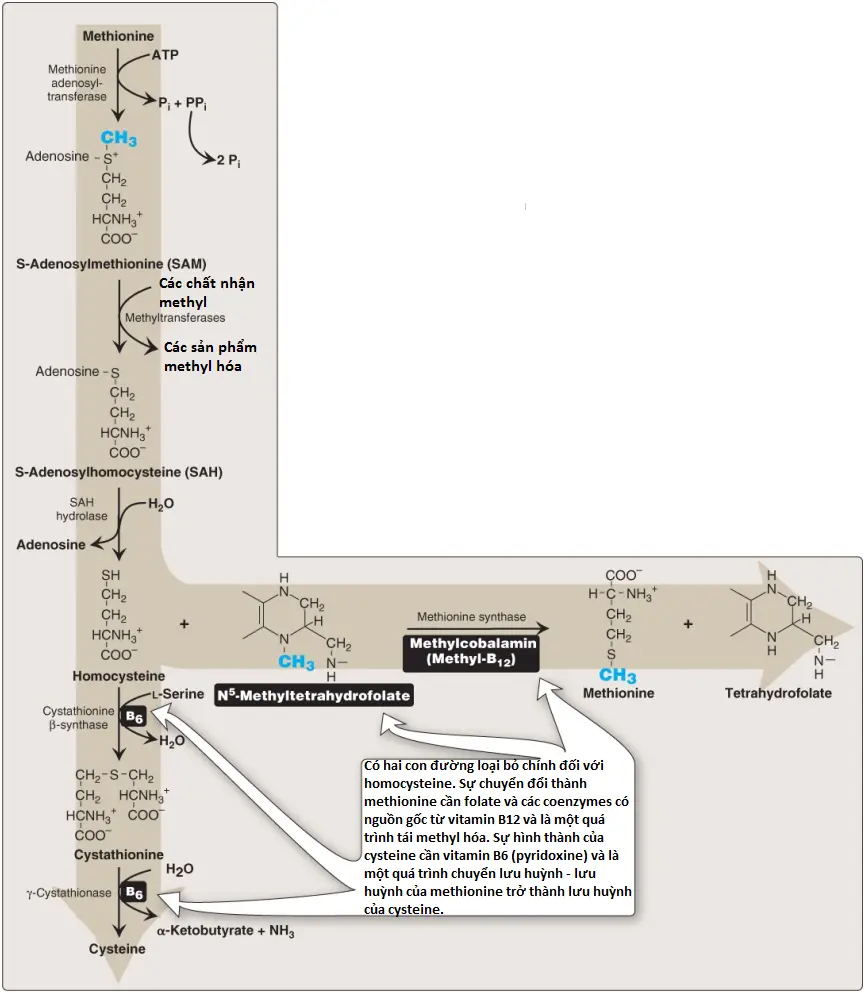
1. Sự tổng hợp S-adenosylmethionine: Methionine ngưng tụ với ATP, hình thành nên SAM, một hợp chất cao năng không thường gặp mà trong đó nó không chứa phosphate. Sự hình thành của SAM được điều khiển bởi sự thủy phân của tất cả ba liên kết phosphate trong ATP (xem Hình 8).
2. Nhóm methyl hoạt hóa: Nhóm methyl liên kết với lưu huỳnh trong SAM được hoạt hóa và có thể được chuyển bởi methyltransferases đến các chất nhận khác nhau như norepinephrine trong sự tổng hợp của epinephrine. Nhóm methyl thường được chuyển đến nitrogen (nitơ) hay các nguyên tử oxygen (như tương ứng lần lượt với sự tổng hợp và thoái hóa epinephrine; xem các bài viết sau) và đôi khi là đến các nguyên tử carbon (như với cytosine). Sản phẩm phản ứng, S-adenosylhomocysteine (SAH), là một thioester đơn giản, tương tự với methionine. Sự mất năng lượng tự do cuối cùng khiến sự chuyển methyl về bản chất là không thể đảo ngược.
3. Sự thủy phân S-adenosylhomocysteine: Sau khi cho nhóm methyl, SAH được thủy phần thành Hcy và adenosine. Hcy có hai số phận. Nếu như có một sự thiếu hụt của methionine, Hcy có thể được tái methyl hóa thành methionine (xem Hình 8). Nếu như các sự tích trữ methionine là đủ thì Hcy có thể đi vào con đường chuyển lưu huỳnh, nơi mà nó được chuyển thành cysteine.
a. Sự tái tổng hợp methionine: Hcy nhận một nhóm methyl từ N5-methyltetrahydrofolate (N5-methyl-TFH) trong một phản ứng cần methylcobalamin, một coenzyme có nguồn gốc từ vitamin B12. (Chú ý: Nhóm methyl được chuyển bởi methionine synthase từ dẫn xuất B12 đến Hcy, tái tạo lại methionine. Cobalamin được tái methyl hóa từ N5-methyl-TFH).
b. Sự tổng hợp cysteine: Được xúc tác bởi cystathionine β-synthase, Hcy ngưng tụ với serine, hình thành nên cystathionine, là chất được thủy phân thành α-ketobutyrate và cysteine (xem Hình 8). Trình tự phản ứng cần vitamin B6 này có một tác động toàn phần là chuyển serine thành cysteine và Hcy thành α-ketobutyrate, là chất được decarboxyl oxy hóa để hình thành nên propionyl CoA. Propionyl CoA được chuyển thành succinyl CoA (xem các bài viết tiếp theo). Bởi vì Hcy được tổng hợp từ amino acid thiết yếu methionine nên cysteine không phải là một amino acid thiết yếu miễn là đủ lượng methionine có sẵn.
4. Mối liên hệ của homocysteine với bệnh mạch máu: Các sự tăng lên trong các mức Hcy huyết tương làm tăng cường tổn thương oxy hóa, viêm và rối loạn chức năng nội mô mạch máu và là một yếu tố nguy cơ độc lập đối với các bệnh mạch máu tắc nghẽn như bệnh tim mạch và đột quỵ (Hình 9). Các sự tăng lên nhẹ (tăng homocysteine máu [hyperhomocysteinemia]) được quan sát thấy ở khoảng 7% dân số. Các nghiên cứu dịch tễ học cho thấy rằng các mức Hcy huyết tương tỷ lệ nghịch với các mức trong huyết tương của folate, B12 và B6, ba vitamin liên quan đến sự chuyển đổi Hcy thành methionine và cysteine. Sự bổ sung các vitamin này được chứng minh là làm giảm các mức Hcy tuần hoàn. Tuy nhiên, ở những bệnh nhân có CVD đã thiết lập, liệu pháp vitamin sẽ không làm giảm các sự kiện tim mạch hay tử vong. Điều này làm tăng lên nghi vấn là liệu Hcy có là nguyên nhân của tổn thương mạch máu hay chỉ đơn thuần là một chỉ điểm cho tổn thương. (Chú ý: Các sự tăng lên nhiều trong mức Hcy huyết tương do các sự thiếu hụt hiếm gặp của enzyme cystathionine β-synthase của con đường chuyển lưu huỳnh được quan sát thấy ở những bệnh nhân mắc homocysteine niệu thể cổ điển [do tăng homocysteine máu nghiêm trọng (>100 micromol/l)]). Các sự thiếu hụt trong phản ứng tái methyl hóa cũng gây ra sự tăng lên trong mức Hcy.
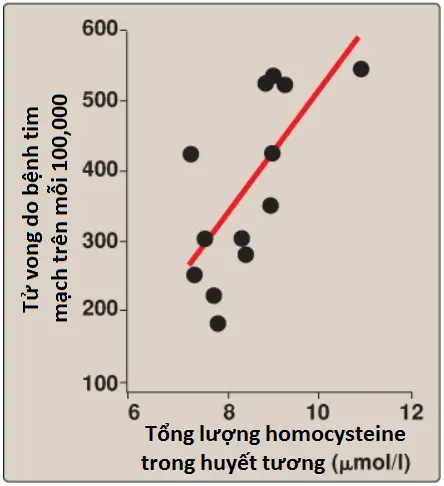
Ở phụ nữ mang thai, các mức Hcy tăng lên thường chỉ điểm cho một sự thiếu hụt trong folic acid, điều này liên quan với một nguy cơ tăng lên của các khiếm khuyết ống thần kinh (sự đóng ống thần kinh không bình thường, như trong dị tật nứt đốt sống) ở thai nhi. Sự bổ sung folate quanh thời điểm mang thai sẽ làm giảm nguy cơ của các khiếm khuyết như vậy.
Các bạn có thể xem bài viết mới trên Facebook tại đây: https://www.facebook.com/profile.php?id=61550892771585
Các bạn có thể xem bài viết trước tại đây: https://docsachxyz.com/amino-acids-su-loai-bo-nitrogen-nito-phan-4/
Cảm ơn các bạn đã theo dõi bài viết. Hẹn gặp lại các bạn trong các bài viết tiếp theo nhé !!!







{kind=link}